|
|
|
|
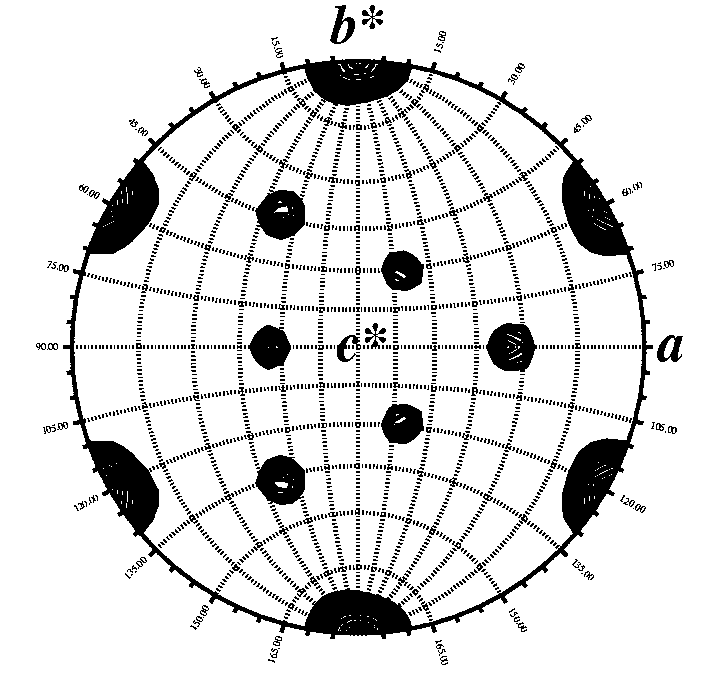
Calculated with GLRF. Map courtesy of S. Lee. |
The self rotation
function is easy to perform. The only requirements are: the crystal's unit
cell parameters, your spacegroup operators, and your data set output by
scalepack.
I typically use the program GLRF from the Replace suite of programs written by Liang Tong. The program produces a contoured map in postscript format that is easily interpreted. An example of a typical input file is given below. Cut and paste the script into a file called self.com. Edit it for your crystal. Execute the file by typing "self.com". View the file "self.ps" with xpsview, showps, display, gs, or whatever postscript interpreter you like. View the sections of Kappa. Manual for GLRF:
Reference for GLRF:
Reference for the Self Rotation Function:
|
|
|
|
|
#!/bin/csh -f /joule2/programs/bin/glrf <<EOF ! Title the Self Rotation Function calculation print self.prt ! !Conventions ! polar xyk euler zyz orthogonalization axabz ! ! ! Crystal A ! acell 52.4 52.4 105.3 90.0 90.0 120.0 asymmetry x, y, z asymmetry -y, x-y, z asymmetry -x+y,-x,z asymmetry -y,-x,-z asymmetry -x+y,y,-z asymmetry x,x-y,-z aobs-file scalepack.sca
|
Important: delete the header from your scalepack file.
self.prt is the log file. It contains a list of the peaks found in the self rotation function. Use these conventions; they are consistent with those used by CCP4 programs.
unit cell parameters for your crystal
symmetry operations of your crystal's space group
WITHOUT fractional
translations! The space group in this example is P3112.
aobs-file. Name of the file containing your scalepack data (intensities
not structure factors in this example)
resolution. You should experiment with the resolution range. (e.g. 9-4, 9-5, 8-4, etc.) If your molecules are relatively floppy, peaks in the RF may only be seen at lower resolution. radius of integration. Again, experiment with this parameter. Use values between the radius and diameter of your protein molecule. slimits. Let phi, psi, and kappa vary from 0 to 180. Use 5 degree
intervals
cntlevel. Contour all peaks above the 450 level, where the highest
peak is scaled to the 1000 level. Show intervals of 50.
|