|
|
|
|
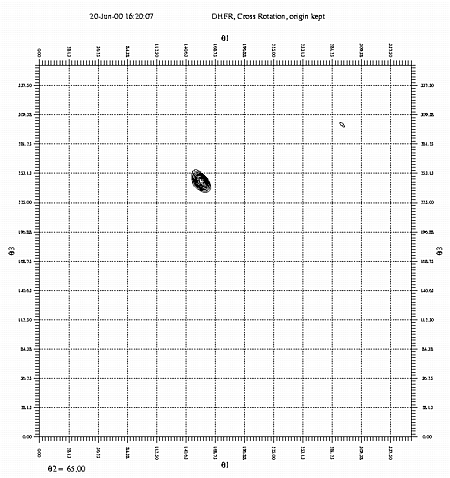 A section of the cross rotation function, Theta2=65. Calculated with GLRF. This example uses data from a crystal of dihydrofolate reductase (DHFR). If you wish to run the example yourself, you may copy the data files from ~sawaya/replace/dhfr/work. The command files may be cut and pasted from the examples below. |
The most straightforward method to calculate the conventional
cross rotation function is to use GLRF from the Replace suite of programs
written by Liang Tong. GLRF uses the Crowther fast rotation function algorithm.
The program produces a contoured map in postscript format that is easily
interpreted. It is fast and intuitive. Additionally, if your asymmetric
unit contains multiple copies of a molecule related by rotational symmetry,
the answers from the cross rotation function in GLRF can be constrained to
have the relationship specified in the self
rotation function. Including this information should improve your
signal to noise ratio. To perform the calculation, you need a
structure factor file for your crystal, and a calculated structure factor
file for your model (in space group P1). Follow the examples below.
Manual for GLRF: Reference for GLRF: Reference for the Crowther Fast Rotation
Function: |
STEP ONE
To calculate structure factors for your model, cut and paste the script below
into a separate file called sfcal.com. Execute the file by typing "sfcal.com"
|
|
|
| #!/bin/csh -f
tf <<EOF title SF calc for DHFR ! print t1.prt cell 100 100 100 90 90 90 ! ! P1 ! symm p1 ! polar xyk euler zyz ortho axabz ! model 1.0 1.0 1.0 90. 90. 90.0 coor format pdb coor type oa coor input dhfr_model.pdb coor flag t coor cen ! ! ! write out pdb file as search model ! coor ou t1o.pdb ! func sfcal t1.cal resolution 500 2.5 direct false table fal stop EOF |
Call the program "tf" to read the following script. t1.prt. Log file for this calculation. symm. Always use space group P1. Use these conventions, they are consistent with CCP4. model. Leave these as they are. Output coordinates of translated molecule. func sfcal. Output calculated structure factor file.
|
STEP TWO
|
Cross Rotation Function with GLRF |
|
| #!/bin/csh -f
glrf <<EOF title DHFR, Cross Rotation, origin kept ! print cross.prt ! ! ! conventions ! polar xyk euler zyz orthogonalization axabz ! ! !locsymmetry 80. 90. 180.0 2 P !locexpand true ! ! acell 100 100 100 90 90 90 asymm x, y, z aobsfile work/t1.cal aformat 3i4, f8.2, 8x, f8.2 acutoff 1 1 0 apower 2 origin false ! bcell 41.580 74.180 59.180 90.00 99.26 90.00 bsymmetry P21 bobsfile work/r1rb3sf.fin bformat (3i4, 2f8.2) bcutoff 2.0 1.0 0.0 bpower 2 ! cutoff 1.5 ! resolution 9 2.5 radius 25.0 ! self false cross true fast true ! sangle euler slimit 1 0 360 5 slimit 2 0 90 5 slimit 3 0 360 5 ! mapfile cross.map peak 2.0 50 oangle polar xyk ! cntfile cross.ps cntlevel 450 1000 50 section 213 cntsection 1 37 ! stop EOF |
Call the program "glrf" to read the following script cross.prt is the log file. It will contain a list of the peaks found in this cross rotation function. Use these conventions; they are consistent with those
used by CCP4 programs. If the self rotation function indicates NCS rotational symmetry, you can specify the angle here. Top solutions in the cross rotation will be constrained to maintain these relationships. Remove the "!" to uncomment. In this example, (80, 90, 180) corresponds to phi, psi, and kappa. "2" specifies a 2-fold axis. "P" specifies that you are using polar angle conventions here. unit cell parameters used to calculate
structure factors from the model (in sfcal.com). unit cell parameters of your crystal.
resolution. You should experiment with the resolution range. (e.g. 9-3, 9-5, 8-4, etc.) If your model is relatively poor, peaks in the RF may only be seen at lower resolution. radius of integration. Again, experiment with this parameter. Use values between the radius and diameter of your protein molecule. slimits. Let theta1, theta2, and theta3 vary over the asymmetric unit of rotational space. Use 5 degree intervals. If you dont know the limits of the asymmetric unit in rotation space, see Rao et al., Acta Cryst. A36, 878-884 (1980). mapfile. The output map containing
the cross rotation function. cntlevel. Contour all peaks above the 450 level, where
the highest peak is scaled to the 1000 level. Show intervals of 50.
|