UCLA Department of Chemistry and Biochemistry
153AH - Fall 2009 - Instructors: Todd Yeates, Duilio Cascio, Tobias Sayre
ATPases: Na+/K +pump (E2-2K+•Pi configuration)
by Daniel Kim
|
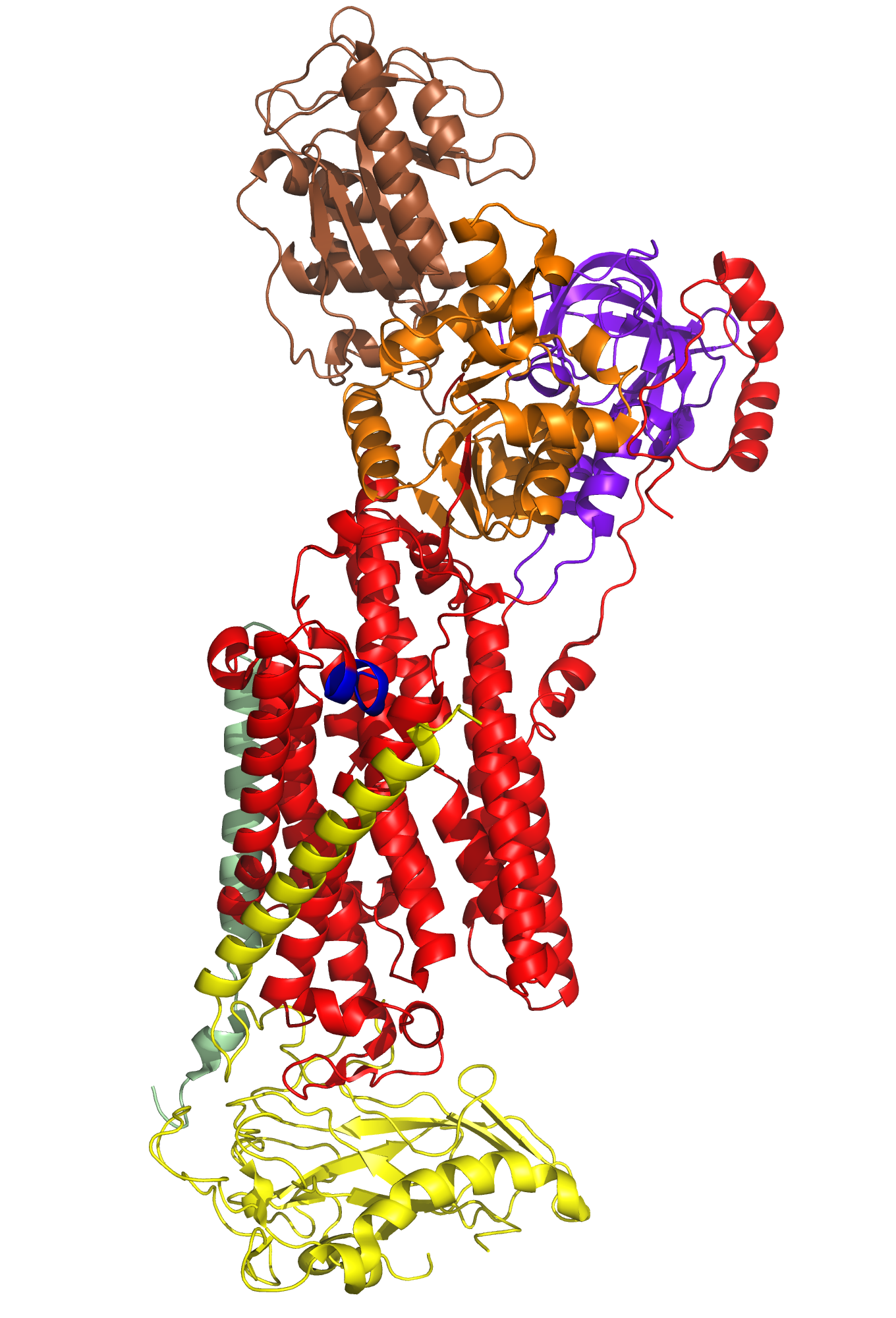
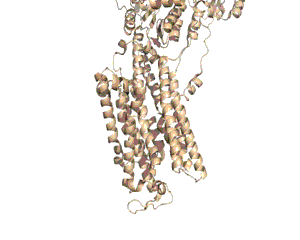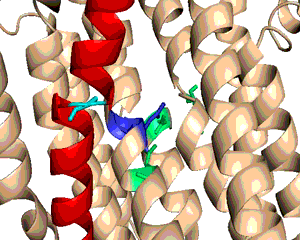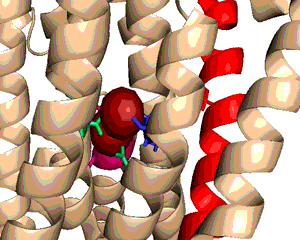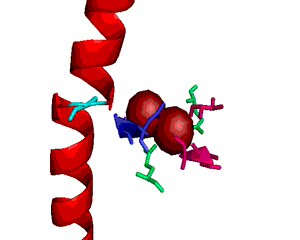
A typical phospholipid bilayer of animal cells contains essential proteins for the transport of nutrients into and out of the cytoplasm to initiate cellular function and regulate cell communication. ATPases are particular types of transmembrane proteins that pump ions against their concentration gradients for special purposes such as creating an action potential for triggering cellular activities. Varied ATPases have shared structural features related to their common functions in binding and hydrolyzing ATP. Na+, K+ ATPases have specific structural features that distinguish them from other ATPases. The binding site for ATP varies between ATPases due to dissimilar interaction between protein domains. Sequence variation and additional subunits explain Na+,K+ ATPase specificity and uniqueness compared to other ATPases such as Ca2+ ATPase. Mutagenesis experiments have determined the effect of particular residues on the protein's binding affinity to sodium and potassium ions. The structural domains located in the cytoplasm and conserved among ATPases are the Actuator, Nucleotide, and Phosphorylation domains (Fig.1). Their varied interactions affect the selectivity towards binding specific ions and the overall ATP turnover rate (2). Specifically, in Na+,K+ ATPases the A and N domains barely interact except through one crucial salt bridge (Fig.2) formed between the positively charged residue Arg551 on the N Domain and the negatively charged residue Glu223 on the A Domain, which aids in ATP binding (3). Furthermore, the P domain in Na+,K+ ATPases positions itself 22° further away from the A and N Domains compared to Ca2+ ATPases, which increases the ATP turnover rate 10-fold (3). Na+,K+ ATPases have unique structural and sequential features that allow only sodium and potassium ions to bind and be transported across a cellular membrane. Among the 10 transmembrane α-helices (Fig.3), one particular helix, αM7, contains a Gly855 residue that distorts the overall helix (3). This distinct kink (absent in Ca2+ ATPases) allows nearby helices to unwind to increase the area of the potassium coordination sites, which involve polar or charged residues containing electronegative oxygen atoms (Fig.3). In addition, the β-subunit, which is located on the extracellular side of the membrane, is another special feature of Na+,K+ ATPases absent in Ca2+ ATPases (Fig.1). It interacts with neighboring α-subunit transmembrane helices through hydrogen bonding to stabilize the binding of K+ ions. Its overall flexibility allows it to act as a mobile lid on the membrane's extracellular side to facilitate and regulate ion transport (3). Nevertheless, in this particular E2-2K+•Pi configuration, affinity for Na+ ions is minimal due to the coordination site's increased size and electrostatically weakened composition (3). Analogous to the β-subunit's role in K+ binding, the C-Terminus (blue) of the α-subunit (Fig.1) regulates Na+ binding by inserting tyrosine residues (after potassium coordination site expansion) to re-distort the αM7 helix to specifically bind Na+(1). The final distinguishing characteristic of the Na+,K+ ATPase is the FXYD motif (Fig.1), contained in the γ-subunit (1), which plays an extracellular regulatory role by serving as an anchor in the membrane and as a hydrogen-bond donor to neighboring helices to stabilize the protein when ions are bound(3). References (1) Morth, J.P., et al. (2007) Crystal structure of the sodium-potassium pump. Nature 450, 1111-1114. (2) Olesen, C., et al. (2007) The structural basis of calcium transport by the calcium pump. Nature 450, 1036-1044. (3) Shinoda, T., et al. (2009) Crystal structure of the sodium-potassium pump at 2.4 Å resolution. Nature 459, 446-451. (4) PDB ID 27XE |
|