UCLA Department of Chemistry and Biochemistry
153AH - Fall 2009 - Instructors: Todd Yeates, Duilio Cascio, Tobias Sayre
XPD Helicase: The Unwinding of Double-Stranded DNA
by Sean O'Connor
|
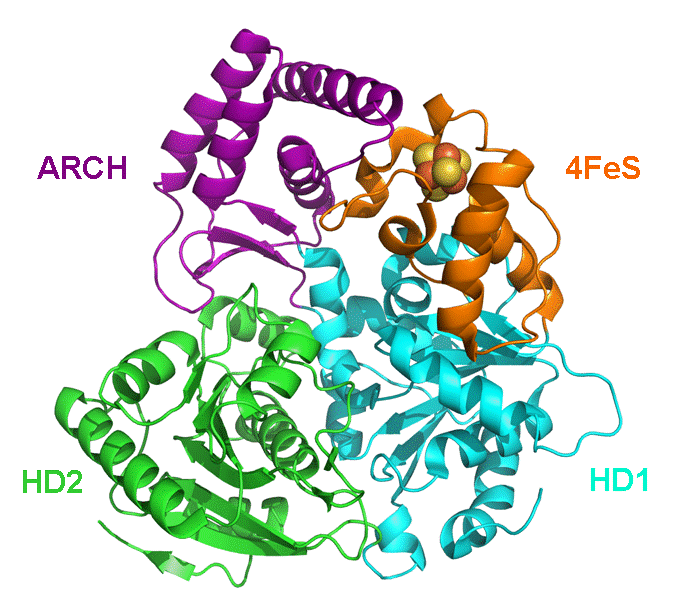
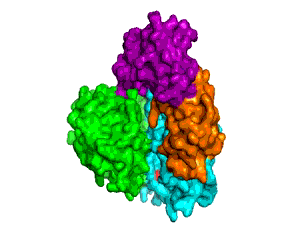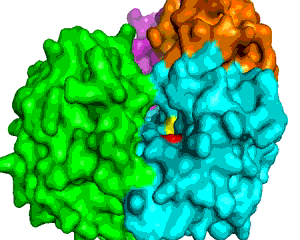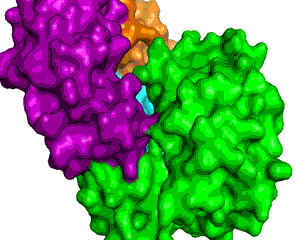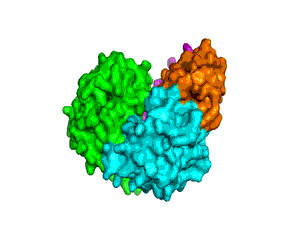
XPD is one of seven protein groups categorizing the autosomal recessive disease Xeroderma pigmentosum (XP) that predisposes individuals to skin cancer (1). However, at least two other autosomal recessive diseases, Cockayne syndrome (CS) and Trichothiodystrophy (TTD) are also associated with XPD (1). In contrast to XP patients, CS and TTD patients are not predisposed to cancer. CS indications include short stature with neurological problems, whereas TTD patients show reductions in sulfur proteins with mental and physical retardation. Biochemically, XPD is a DNA helicase that unwinds DNA at the expense of ATP. XPD participates in the RNA Polymerase II subunit TFIIH complex that has a role in both transcription and nucleotide excision repair (NER). XPD is homologous to S. cerevisiae Rad3 and, in addition to the ATPase, has a 4Fe4S cluster (2). In the 1990s, a connection between different sequence mutations in XPD and the different clinical disease manifestations was established (1). Recently, a molecular understanding of how different mutations in the same protein can lead to different diseases was provided by the crystal structure of an XPD homolog from Sulfolobus acidocaldarius (SaXPD) (Figure 1) (3). SaXPD was separated into four domains, an Arch, Helicase Domains 1 and 2 (HD1 and HD2), and the 4FeS domain (Figure 1) (3). In the process of unwinding DNA, a single strand of DNA binds in a pocket on the surface and is threaded, 10 nucleotides at a time, through the arch by a positively charged electrostatic 'tunnel' (Figure 2) (3). Mutations altering the single-strand DNA binding domain result in XP. Threading of the single-strand of DNA requires ATP hydrolysis, and the ATP binding region is located between the HD1 and HD2 domains (3). Alterations in the flexibility of the HD1 and HD2 domains are associated with XP/CS, whereas TTD is linked to mutations that change the overall SaXPD structural framework. While three specific disease mutations in the vicinity of the ATP binding groove are directly associated with the absence of ATP hydrolysis and protein activity, ATP usage is still sensitive to most other mutations in the HD1 and HD2 regions (Figure 2 and 3) (3). The 4Fe4S cluster is composed of four helices linked by loops and stabilized by the four cysteine ligand interactions with Fe atoms (3). The 4FeS domain is critical to the maintenance of structural integrity since mutation or removal of the iron-sulfur ligand complex results protein unfolding with concomitant loss of protein activity (3). Comparing the SaXPD and human XPD domains shows that different disease phenotypes are based on whether DNA binding, ATPase activity, or helicase structure are affected (3). Understanding implications of SaXPD mutations can also provide data on key structural elements needed to maintain interactions with other proteins in large complexes involved in transcription and DNA repair. Future research will investigate the interaction of SaXPD and XPD features in their larger molecular environment and thus how mutations, acting alone or in concert, can translate to various pathologies (3). References 1. Taylor, E. M., Broughton, B. C., Botta, E., Stefanini, M., Sarasin, A., Jaspers, N. G., Fawcett, H., Harcourt, S. A., Arlett, C. F., and Lehmann, A. R. (1997) Xeroderma pigmentosum and trichothiodystrophy are associated with different mutations in the XPD (ERCC2) repair/transcription gene. Proc Natl Acad Sci USA 94, 8658-8663. 2. Guzder, S. N., Qiu, H., Sommers, C. H., Sung, P., Prakash, L., and Prakash, S. (1994) DNA repair gene RAD3 of S. cerevisiae is essential for transcription by RNA polymerase II. Nature 367, 91-94. 3. Fan, L., Fuss, J. O., Cheng, Q. J., Arvai, A. S., Hammel, M., Roberts, V. A., Cooper, P. K., and Tainer, J. A. (2008) XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell 133, 789-800. 4.PDBID 3crv |
|