UCLA Department of Chemistry and Biochemistry
153AH - Fall 2009 - Instructors: Todd Yeates, Duilio Cascio, Tobias Sayre
HIV-1 Protease: A Target for AIDS Therapy
by Pathu Sriphanlop
|
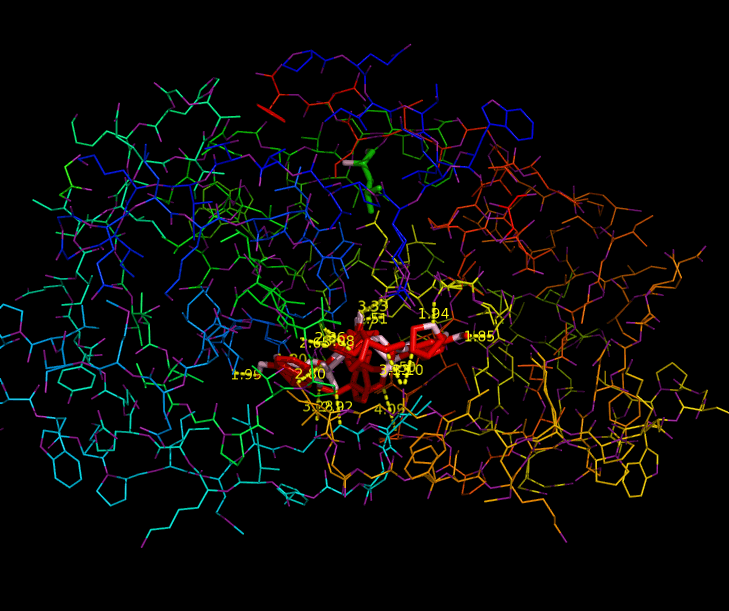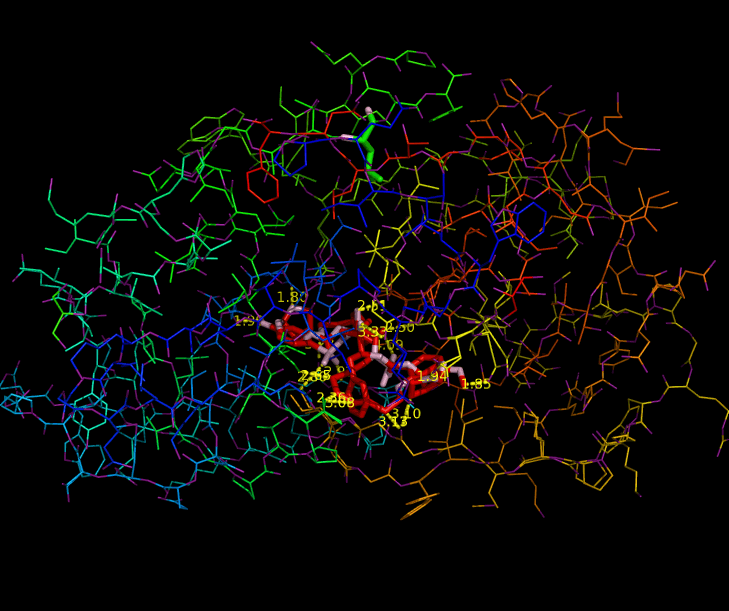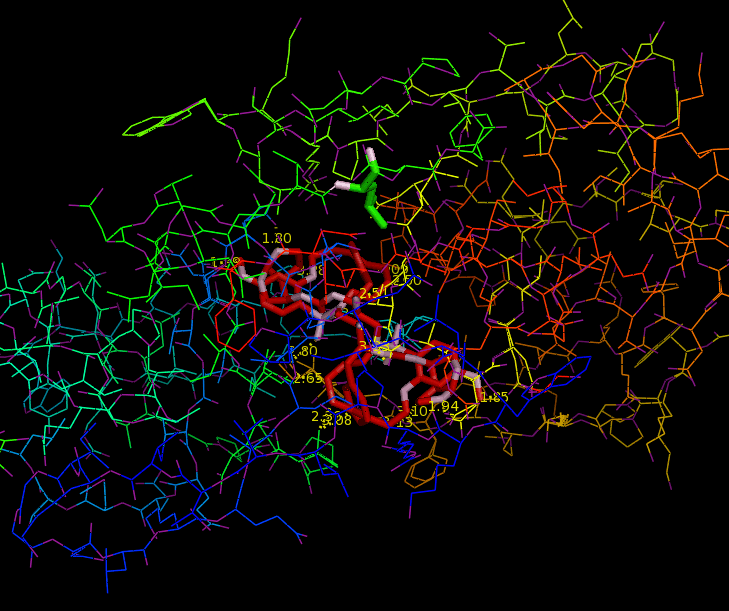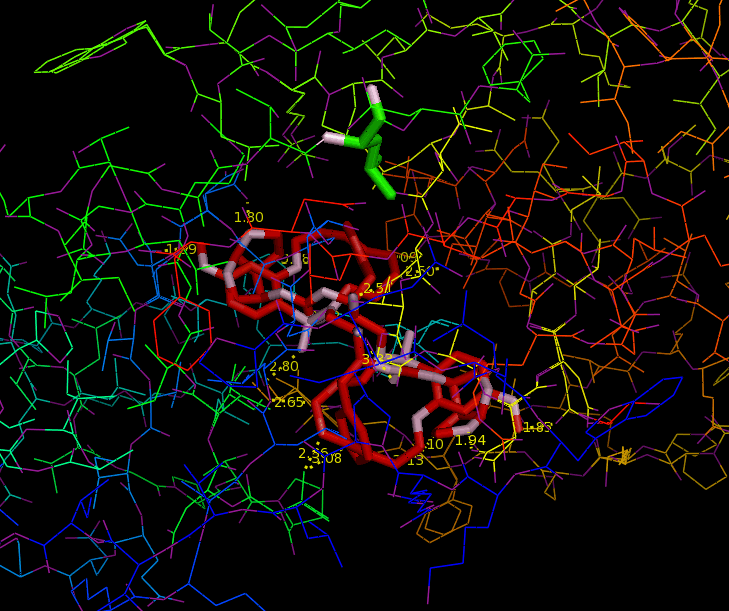
Since HIV protease inhibitors were first introduced in 1995, they have greatly benefited those infected by HIV by prolonging viral control, suppressing the virus, and reducing mortality. HIV-1 Protease plays a major role in the life cycle of HIV. Because HIV makes its proteins in one long piece, HIV-1 Protease must cut this polyprotein into smaller protein-sized pieces. To form the mature virus that will then infect a new cell, this cleavage must occur after the immature virus has assembled properly. Due to its time-sensitivity and crucial role, HIV-1 Protease is an effective target for drug therapy. Competitive inhibitors are used to bind to the protease and block its function, thereby suppressing the virus, which cannot transform to its mature, infectious form (2). The structure of HIV-1 Protease has been extensively studied for the purpose of drug design. The Protease has flaps that cover the top of the active site. Successful inhibitors tend to be carbon-rich groups stretched straight through the active site, thereby interacting with the sides of the active site tunnel (Fig. 1). These inhibitors mimic a protein chain and therefore bind tightly to the enzyme. HIV-1 Protease cannot cleave them, so they remain bound to the active site and block its normal function. New protease inhibitors are continuously studied and designed today because resistance to them has become a major limitation to drug therapy. Thermodynamic studies show that the hydrophobic effect was the major driving force for binding earlier inhibitors (older drugs). Because these inhibitors had relatively rigid shapes, they could not adapt to mutations in the target enzyme. More recent inhibitors are able to maintain higher affinities despite mutations in the protease sequence. This shows that effective drugs need to have high affinity and high adaptability to mutations (2). Studies aimed at developing inhibitors that treat inhibitor-resistant HIV strains demonstrate that inhibitors must fit the expanded active site of the drug-resistant proteases. Therefore, protease inhibitors must be adaptive and fill the expanded active site cavity due to mutation. Ghosh et al. studied different inhibitor designs that best combat drug resistance. The structures of protein-ligand complexes showed extensive hydrogen bonding interactions with the backbone atoms throughout the active site. Therefore, they concluded that maximizing "backbone binding" is an important design strategy to combat drug resistance. Another notable trend in inhibitors designed to combat drug resistance is a macrocyclic structure, which increases the size of the hydrophobic pocket. They also showed that elongating the carbon chain resulted in lower enzyme inhibitory activity. In addition, converting acyclic inhibitors to their cyclic analogues improved enzyme inhibitory and antiviral activity; the cyclic analogues make extensive hydrogen bonds with the protease backbone and effectively fill the hydrophobic pockets (1). References (1) Ghosh, et al. (2009). Design, Synthesis, Protein-Ligand X-ray Structure, and Biological Evaluation of a Series of Novel Macrocyclic Human Immunodeficiency Virus-1 Protease Inhibitors to Combat Drug Resistance. J. Med. Chem. 52, 7689-705. (2) Martinez-Cajas, et al. (2007). Protease inhibitor resistance in HIV-infected patients: Molecular and clinical perspectives. Antiviral Res. 76, 203-221. (3) PDBID 2IEN, 3I7E, 3I6O |
|