|
The genome revolution has produced an abundance of protein
sequence data. Traditional homology-based computer methods make
it possible to establish evolutionary relationships between large
numbers of these proteins. Yet among any set of new protein
sequences,
say from the complete genome sequence of a new organism, a
significant fraction of the proteins cannot be
assigned fuctions by traditional methods. A new sequence may
have no recognizable homologs in other
organisms, or it may have recognizable homologs, the cellular
functions of which are yet unknown. The critical need
to make some kind of functional inferences for the vast numbers
of proteins that could not be functionally annoted by traditional
homology methods led in 1999, and in the years that followed,
to new ideas for inferring 'functional
linkages' between proteins not related to each other by
homology. These 'non-homology' or 'genomic context' methods
included the 'Phylogenetic Profile' method and the 'Rosetta-Stone'
method (both pioneered principally by Edward Marcotte and Matteo Pellegrini
when they were postdocs with Eisenberg and Yeates), and others.
Subsequent work has aimed to extend those
ideas. One recent extension of Phylogenetic Profiles
(developed by Peter Bowers and Shawn Cokus)
involves an application of logic
analysis to uncover proteins whose presence vs. absence across
organisms is related to the presence or absence of two other
proteins, taken in logical combination. These kinds of higher
order relationships are
expected to be abundant in the cell, but are not detected
by the original Phylogenetic Profile method, which looks for
direct similarity between the profiles of just two proteins
at a time.
Our computational genomics work has touched on many other subjects as well: disulfide bonding in thermophiles,
repetitive protein sequences, genomic encoding of unusual amino acids such as selenocysteine and pyrollysine,
detection of protein targeting sequences, and the function of bacterial microcompartments.
References:
- Jorda J, Yeates TO. (2011).
Widespread disulfide bonding in proteins from thermophilic archaea.
Archaea. 2011. 2011:409156.
[Abstract]
Disulfide bonds are generally not used to stabilize proteins in the cytosolic compartments of bacteria or eukaryotic cells, owing to the chemically reducing nature of those environments. In contrast, certain thermophilic archaea use disulfide bonding as a major mechanism for protein stabilization. Here, we provide a current survey of completely sequenced genomes, applying computational methods to estimate the use of disulfide bonding across the Archaea. Microbes belonging to the Crenarchaeal branch, which are essentially all hyperthermophilic, are universally rich in disulfide bonding while lesser degrees of disulfide bonding are found among the thermophilic Euryarchaea, excluding those that are methanogenic. The results help clarify which parts of the archaeal lineage are likely to yield more examples and additional specific data on protein disulfide bonding, as increasing genomic sequencing efforts are brought to bear.
- Fan C, Cheng S, Liu Y, Escobar CM, Crowley CS, Jefferson RE, Yeates TO, Bobik TA. (2010).
Short N-terminal sequences package proteins into bacterial microcompartments.
Proc. Natl. Acad. Sci. U.S.A.. Apr 2010. 107(16):7509-14.
[Abstract]
Hundreds of bacterial species produce proteinaceous microcompartments (MCPs) that act as simple organelles by confining the enzymes of metabolic pathways that have toxic or volatile intermediates. A fundamental unanswered question about bacterial MCPs is how enzymes are packaged within the protein shell that forms their outer surface. Here, we report that a short N-terminal peptide is necessary and sufficient for packaging enzymes into the lumen of an MCP involved in B(12)-dependent 1,2-propanediol utilization (Pdu MCP). Deletion of 10 or 14 amino acids from the N terminus of the propionaldehyde dehydrogenase (PduP) enzyme, which is normally found within the Pdu MCP, substantially impaired packaging, with minimal effects on its enzymatic activity. Fusion of the 18 N-terminal amino acids from PduP to GFP, GST, or maltose-binding protein resulted in their encapsulation within MCPs. Bioinformatic analyses revealed N-terminal extensions in two additional Pdu proteins and three proteins from two unrelated MCPs, suggesting that N-terminal peptides may be used to package proteins into diverse MCPs. The potential uses of MCP assembly principles in nature and in biotechnology are discussed.
- Sprinzak E, Cokus SJ, Yeates TO, Eisenberg D, Pellegrini M. (2009).
Detecting coordinated regulation of multi-protein complexes using logic analysis of gene expression.
BMC Syst Biol. 2009. 3:115.
[Abstract]
Many of the functional units in cells are multi-protein complexes such as RNA polymerase, the ribosome, and the proteasome. For such units to work together, one might expect a high level of regulation to enable co-appearance or repression of sets of complexes at the required time. However, this type of coordinated regulation between whole complexes is difficult to detect by existing methods for analyzing mRNA co-expression. We propose a new methodology that is able to detect such higher order relationships.
- Beeby M, Bobik TA, Yeates TO. (2009).
Exploiting genomic patterns to discover new supramolecular protein assemblies.
Protein Sci.. Jan 2009. 18(1):69-79.
[Abstract]
Bacterial microcompartments are supramolecular protein assemblies that function as bacterial organelles by compartmentalizing particular enzymes and metabolic intermediates. The outer shells of these microcompartments are assembled from multiple paralogous structural proteins. Because the paralogs are required to assemble together, their genes are often transcribed together from the same operon, giving rise to a distinctive genomic pattern: multiple, typically small, paralogous proteins encoded in close proximity on the bacterial chromosome. To investigate the generality of this pattern in supramolecular assemblies, we employed a comparative genomics approach to search for protein families that show the same kind of genomic pattern as that exhibited by bacterial microcompartments. The results indicate that a variety of large supramolecular assemblies fit the pattern, including bacterial gas vesicles, bacterial pili, and small heat-shock protein complexes. The search also retrieved several widely distributed protein families of presently unknown function. The proteins from one of these families were characterized experimentally and found to show a behavior indicative of supramolecular assembly. We conclude that cotranscribed paralogs are a common feature of diverse supramolecular assemblies, and a useful genomic signature for discovering new kinds of large protein assemblies from genomic data.
- Chaudhuri BN, Yeates TO. (2005).
A computational method to predict genetically encoded rare amino acids in proteins.
Genome Biol.. 2005. 6(9):R79.
[Abstract]
In several natural settings, the standard genetic code is expanded to incorporate two additional amino acids with distinct functionality, selenocysteine and pyrrolysine. These rare amino acids can be overlooked inadvertently, however, as they arise by recoding at certain stop codons. We report a method for such recoding prediction from genomic data, using read-through similarity evaluation. A survey across a set of microbial genomes identifies almost all the known cases as well as a number of novel candidate proteins.
- Beeby M, O'Connor BD, Ryttersgaard C, Boutz DR, Perry LJ, Yeates TO. (2005).
The genomics of disulfide bonding and protein stabilization in thermophiles.
PLoS Biol.. Sep 2005. 3(9):e309.
[Abstract]
Thermophilic organisms flourish in varied high-temperature environmental niches that are deadly to other organisms. Recently, genomic evidence has implicated a critical role for disulfide bonds in the structural stabilization of intracellular proteins from certain of these organisms, contrary to the conventional view that structural disulfide bonds are exclusively extracellular. Here both computational and structural data are presented to explore the occurrence of disulfide bonds as a protein-stabilization method across many thermophilic prokaryotes. Based on computational studies, disulfide-bond richness is found to be widespread, with thermophiles containing the highest levels. Interestingly, only a distinct subset of thermophiles exhibit this property. A computational search for proteins matching this target phylogenetic profile singles out a specific protein, known as protein disulfide oxidoreductase, as a potential key player in thermophilic intracellular disulfide-bond formation. Finally, biochemical support in the form of a new crystal structure of a thermophilic protein with three disulfide bonds is presented together with a survey of known structures from the literature. Together, the results provide insight into biochemical specialization and the diversity of methods employed by organisms to stabilize their proteins in exotic environments. The findings also motivate continued efforts to sequence genomes from divergent organisms.
- Bowers PM, O'Connor BD, Cokus SJ, Sprinzak E, Yeates TO, Eisenberg D. (2005).
Utilizing logical relationships in genomic data to decipher cellular processes.
FEBS J.. Oct 2005. 272(20):5110-8.
[Abstract]
The wealth of available genomic data has spawned a corresponding interest in computational methods that can impart biological meaning and context to these experiments. Traditional computational methods have drawn relationships between pairs of proteins or genes based on notions of equality or similarity between their patterns of occurrence or behavior. For example, two genes displaying similar variation in expression, over a number of experiments, may be predicted to be functionally related. We have introduced a natural extension of these approaches, instead identifying logical relationships involving triplets of proteins. Triplets provide for various discrete kinds of logic relationships, leading to detailed inferences about biological associations. For instance, a protein C might be encoded within an organism if, and only if, two other proteins A and B are also both encoded within the organism, thus suggesting that gene C is functionally related to genes A and B. The method has been applied fruitfully to both phylogenetic and microarray expression data, and has been used to associate logical combinations of protein activity with disease state phenotypes, revealing previously unknown ternary relationships among proteins, and illustrating the inherent complexities that arise in biological data.
- Bowers PM, Pellegrini M, Thompson MJ, Fierro J, Yeates TO, Eisenberg D. (2004).
Prolinks: a database of protein functional linkages derived from coevolution.
Genome Biol.. 2004. 5(5):R35.
[Abstract]
The advent of whole-genome sequencing has led to methods that infer protein function and linkages. We have combined four such algorithms (phylogenetic profile, Rosetta Stone, gene neighbor and gene cluster) in a single database--Prolinks--that spans 83 organisms and includes 10 million high-confidence links. The Proteome Navigator tool allows users to browse predicted linkage networks interactively, providing accompanying annotation from public databases. The Prolinks database and the Proteome Navigator tool are available for use online at http://dip.doe-mbi.ucla.edu/pronav.
- O'Connor BD, Yeates TO. (2004).
GDAP: a web tool for genome-wide protein disulfide bond prediction.
Nucleic Acids Res.. Jul 2004. 32(Web Server issue):W360-4.
[Abstract]
The Genomic Disulfide Analysis Program (GDAP) provides web access to computationally predicted protein disulfide bonds for over one hundred microbial genomes, including both bacterial and achaeal species. In the GDAP process, sequences of unknown structure are mapped, when possible, to known homologous Protein Data Bank (PDB) structures, after which specific distance criteria are applied to predict disulfide bonds. GDAP also accepts user-supplied protein sequences and subsequently queries the PDB sequence database for the best matches, scans for possible disulfide bonds and returns the results to the client. These predictions are useful for a variety of applications and have previously been used to show a dramatic preference in certain thermophilic archaea and bacteria for disulfide bonds within intracellular proteins. Given the central role these stabilizing, covalent bonds play in such organisms, the predictions available from GDAP provide a rich data source for designing site-directed mutants with more stable thermal profiles. The GDAP web application is a gateway to this information and can be used to understand the role disulfide bonds play in protein stability both in these unusual organisms and in sequences of interest to the individual researcher. The prediction server can be accessed at http://www.doe-mbi.ucla.edu/Services/GDAP.
- Bowers PM, Cokus SJ, Eisenberg D, Yeates TO. (2004).
Use of logic relationships to decipher protein network organization.
Science. Dec 2004. 306(5705):2246-9.
[Abstract]
A major focus of genome research is to decipher the networks of molecular interactions that underlie cellular function. We describe a computational approach for identifying detailed relationships between proteins on the basis of genomic data. Logic analysis of phylogenetic profiles identifies triplets of proteins whose presence or absence obey certain logic relationships. For example, protein C may be present in a genome only if proteins A and B are both present. The method reveals many previously unidentified higher order relationships. These relationships illustrate the complexities that arise in cellular networks because of branching and alternate pathways, and they also facilitate assignment of cellular functions to uncharacterized proteins.
- Strong M, Graeber TG, Beeby M, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg D. (2003).
Visualization and interpretation of protein networks in Mycobacterium tuberculosis based on hierarchical clustering of genome-wide functional linkage maps.
Nucleic Acids Res.. Dec 2003. 31(24):7099-109.
[Abstract]
Genome-wide functional linkages among proteins in cellular complexes and metabolic pathways can be inferred from high throughput experimentation, such as DNA microarrays, or from bioinformatic analyses. Here we describe a method for the visualization and interpretation of genome-wide functional linkages inferred by the Rosetta Stone, Phylogenetic Profile, Operon and Conserved Gene Neighbor computational methods. This method involves the construction of a genome-wide functional linkage map, where each significant functional linkage between a pair of proteins is displayed on a two-dimensional scatter-plot, organized according to the order of genes along the chromosome. Subsequent hierarchical clustering of the map reveals clusters of genes with similar functional linkage profiles and facilitates the inference of protein function and the discovery of functionally linked gene clusters throughout the genome. We illustrate this method by applying it to the genome of the pathogenic bacterium Mycobacterium tuberculosis, assigning cellular functions to previously uncharacterized proteins involved in cell wall biosynthesis, signal transduction, chaperone activity, energy metabolism and polysaccharide biosynthesis.
- Mallick P, Boutz DR, Eisenberg D, Yeates TO. (2002).
Genomic evidence that the intracellular proteins of archaeal microbes contain disulfide bonds.
Proc. Natl. Acad. Sci. U.S.A.. Jul 2002. 99(15):9679-84.
[Abstract]
Disulfide bonds have only rarely been found in intracellular proteins. That pattern is consistent with the chemically reducing environment inside the cells of well-studied organisms. However, recent experiments and new calculations based on genomic data of archaea provide striking contradictions to this pattern. Our results indicate that the intracellular proteins of certain hyperthermophilic archaea, especially the crenarchaea Pyrobaculum aerophilum and Aeropyrum pernix, are rich in disulfide bonds. This finding implicates disulfide bonding in stabilizing many thermostable proteins and points to novel chemical environments inside these microbes. These unexpected results illustrate the wealth of biochemical insights available from the growing reservoir of genomic data.
- Eisenberg D, Marcotte EM, Xenarios I, Yeates TO. (2000).
Protein function in the post-genomic era.
Nature. Jun 2000. 405(6788):823-6.
[Abstract]
Faced with the avalanche of genomic sequences and data on messenger RNA expression, biological scientists are confronting a frightening prospect: piles of information but only flakes of knowledge. How can the thousands of sequences being determined and deposited, and the thousands of expression profiles being generated by the new array methods, be synthesized into useful knowledge? What form will this knowledge take? These are questions being addressed by scientists in the field known as 'functional genomics'.
- Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. (1999).
Assigning protein functions by comparative genome analysis: protein phylogenetic profiles.
Proc. Natl. Acad. Sci. U.S.A.. Apr 1999. 96(8):4285-8.
[Abstract]
Determining protein functions from genomic sequences is a central goal of bioinformatics. We present a method based on the assumption that proteins that function together in a pathway or structural complex are likely to evolve in a correlated fashion. During evolution, all such functionally linked proteins tend to be either preserved or eliminated in a new species. We describe this property of correlated evolution by characterizing each protein by its phylogenetic profile, a string that encodes the presence or absence of a protein in every known genome. We show that proteins having matching or similar profiles strongly tend to be functionally linked. This method of phylogenetic profiling allows us to predict the function of uncharacterized proteins.
- Pellegrini M, Marcotte EM, Yeates TO. (1999).
A fast algorithm for genome-wide analysis of proteins with repeated sequences.
Proteins. Jun 1999. 35(4):440-6.
[Abstract]
We present a fast algorithm to search for repeating fragments within protein sequences. The technique is based on an extension of the Smith-Waterman algorithm that allows the calculation of sub-optimal alignments of a sequence against itself. We are able to estimate the statistical significance of all sub-optimal alignment scores. We also rapidly determine the length of the repeating fragment and the number of times it is found in a sequence. The technique is applied to sequences in the Swissprot database, and to 16 complete genomes. We find that eukaryotic proteins contain more internal repeats than those of prokaryotic and archael organisms. The finding that 18% of yeast sequences and 28% of the known human sequences contain detectable repeats emphasizes the importance of internal duplication in protein evolution.
- Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. (1999).
Detecting protein function and protein-protein interactions from genome sequences.
Science. Jul 1999. 285(5428):751-3.
[Abstract]
A computational method is proposed for inferring protein interactions from genome sequences on the basis of the observation that some pairs of interacting proteins have homologs in another organism fused into a single protein chain. Searching sequences from many genomes revealed 6809 such putative protein-protein interactions in Escherichia coli and 45,502 in yeast. Many members of these pairs were confirmed as functionally related; computational filtering further enriches for interactions. Some proteins have links to several other proteins; these coupled links appear to represent functional interactions such as complexes or pathways. Experimentally confirmed interacting pairs are documented in a Database of Interacting Proteins.
- Marcotte EM, Pellegrini M, Yeates TO, Eisenberg D. (1999).
A census of protein repeats.
J. Mol. Biol.. Oct 1999. 293(1):151-60.
[Abstract]
In this study, we analyzed all known protein sequences for repeating amino acid segments. Although duplicated sequence segments occur in 14 % of all proteins, eukaryotic proteins are three times more likely to have internal repeats than prokaryotic proteins. After clustering the repetitive sequence segments into families, we find repeats from eukaryotic proteins have little similarity with prokaryotic repeats, suggesting most repeats arose after the prokaryotic and eukaryotic lineages diverged. Consequently, protein classes with the highest incidence of repetitive sequences perform functions unique to eukaryotes. The frequency distribution of the repeating units shows only weak length dependence, implicating recombination rather than duplex melting or DNA hairpin formation as the limiting mechanism underlying repeat formation. The mechanism favors additional repeats once an initial duplication has been incorporated. Finally, we show that repetitive sequences are favored that contain small and relatively water-soluble residues. We propose that error-prone repeat expansion allows repetitive proteins to evolve more quickly than non-repeat-containing proteins.
- Pellegrini M, Yeates TO. (1999).
Searching for frameshift evolutionary relationships between protein sequence families.
Proteins. Nov 1999. 37(2):278-83.
[Abstract]
The protein sequence database was analyzed for evidence that some distinct sequence families might be distantly related in evolution by changes in frame of translation. Sequences were compared using special amino acid substitution matrices for the alternate frames of translation. The statistical significance of alignment scores were computed in the true database and shuffled versions of the database that preserve any potential codon bias. The comparison of results from these two databases provides a very sensitive method for detecting remote relationships. We find a weak but measurable relatedness within the database as a whole, supporting the notion that some proteins may have evolved from others through changes in frame of translation. We also quantify residual homology in the ordinary sense within a database of generally unrelated sequences.
- Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg D. (1999).
A combined algorithm for genome-wide prediction of protein function.
Nature. Nov 1999. 402(6757):83-6.
[Abstract]
The availability of over 20 fully sequenced genomes has driven the development of new methods to find protein function and interactions. Here we group proteins by correlated evolution, correlated messenger RNA expression patterns and patterns of domain fusion to determine functional relationships among the 6,217 proteins of the yeast Saccharomyces cerevisiae. Using these methods, we discover over 93,000 pairwise links between functionally related yeast proteins. Links between characterized and uncharacterized proteins allow a general function to be assigned to more than half of the 2,557 previously uncharacterized yeast proteins. Examples of functional links are given for a protein family of previously unknown function, a protein whose human homologues are implicated in colon cancer and the yeast prion Sup35.
|
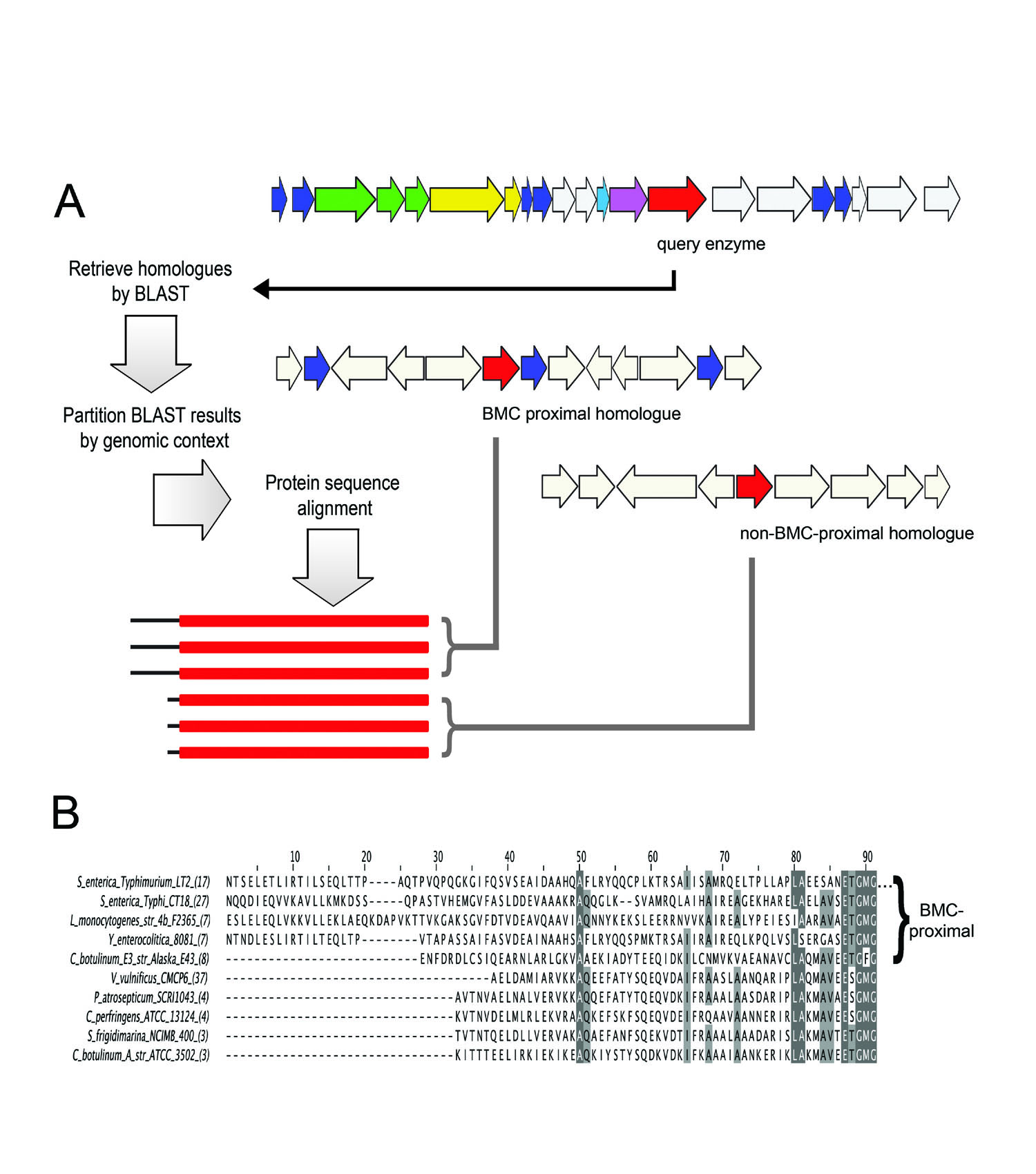
A diagram illustrating a method for predicting mechanisms of protein targeting
(e.g. to bacterial microcompartments) by special N or C-terminal sequence extensions.
(Adapted from Fan, et al. 2010)
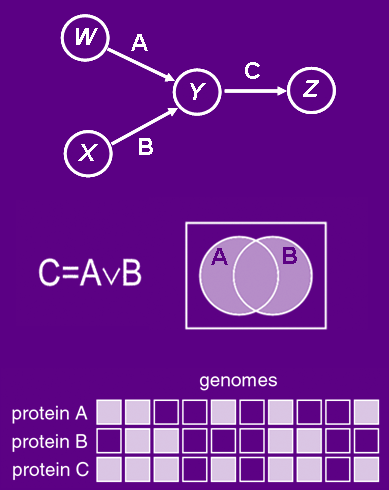
A diagram illustrating the idea of logic analysis of phylogenetic profiles. (Adapted from Bowers, et al. 2002)
|