|
Although proteins often adopt rather complex folds, the fact
of the matter is that the backbone of a protein molecule adopts a
knotted configuration in only
a very few proteins among the thousands of known protein folds. This is
contrary to what would be expected if protein backbones followed essentially
random paths in space; for spatial curves as long as medium-sized
proteins, knots would be expected to dominate. The observation
that native protein folds hardly sample the space of knotted
conformations - a space that represents the majority of all
possible conformations - has profound implications
for protein folding in general, and for the Thermodynamic
Hypothesis (first posed by Anfinsen) in particular.
Despite their rarity, there are a few special cases where
knotted proteins have been observed, and these provide
special opportunities for studying protein folding and stability.
The intuition that it might be kinetically difficult for a
knotted protein chain to reach its native state can be considered
in reverse, with the conclusion that knotting and other kinds
of topological entaglements could kinetically stabilize the
folded states of proteins containing these features. While looking
for unusual topological features in protein stuctures,
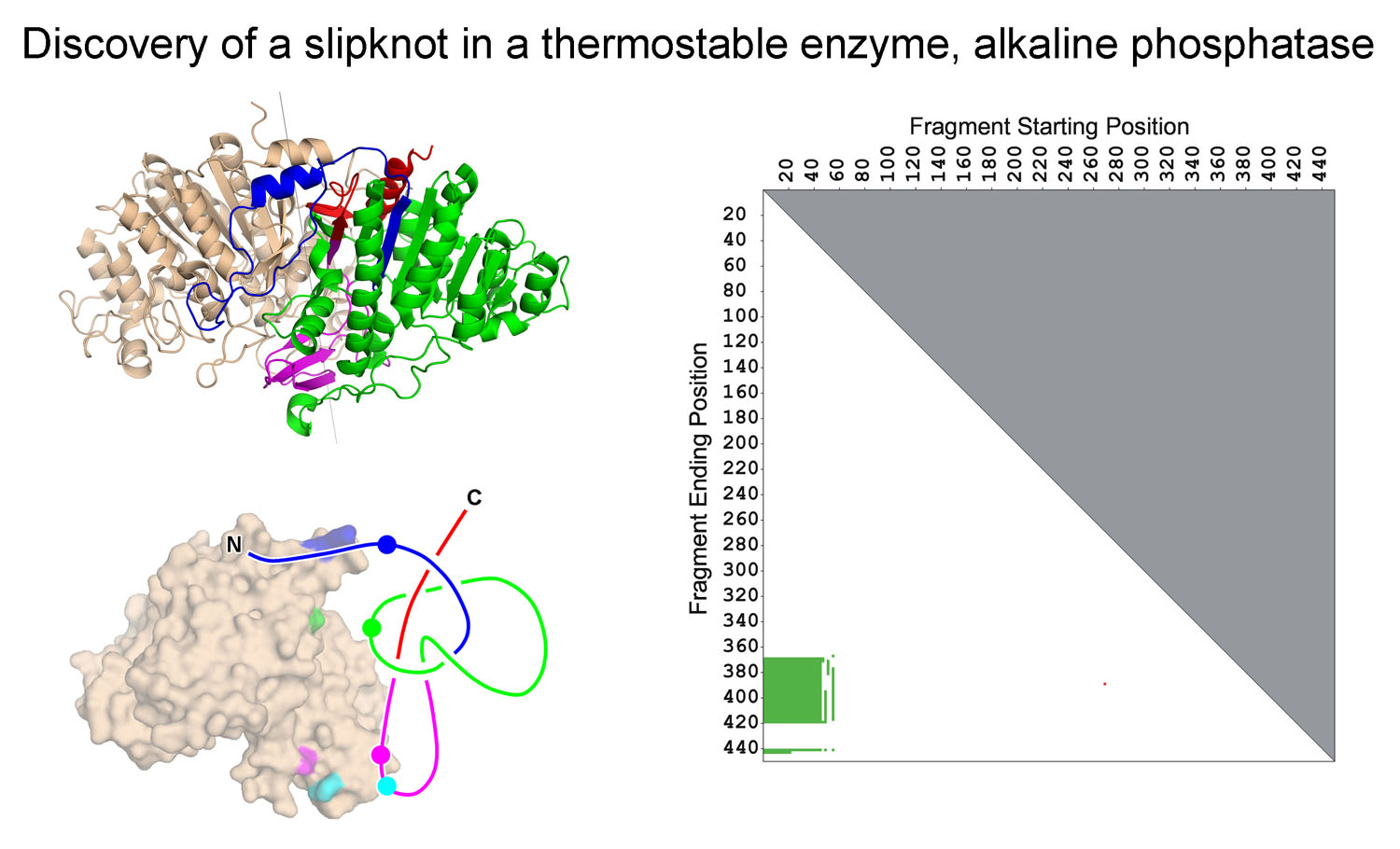
we (Neil King) discovered a novel slipknot feature in a well-known
thermostable protein, alkaline phosphatase. Slipknots in proteins
had eluded previous computational methods that searched protein
structures for knots, since previous programs only analazed a
complete protein backbone in its entirety; when a slipknot is present, the complete curve
appears to be unknotted (since if you pull on the two ends the knot would be undone),
but a true knot exists as a partial structure within the backbone.
Neil's 2007 JMB paper
introduced the idea of a 2D knotplot (see figure second from top) to convey whether
knots were contained as partial structures within proteins.
Beyond the knotting of individual protein chains, a few rare
cases are known in which protein chains from two different
subunits form topologically interlinked chains. One of these
was discovered in the lab by Danny Boutz
in the citrate synthase dimer from P. aerophilum.
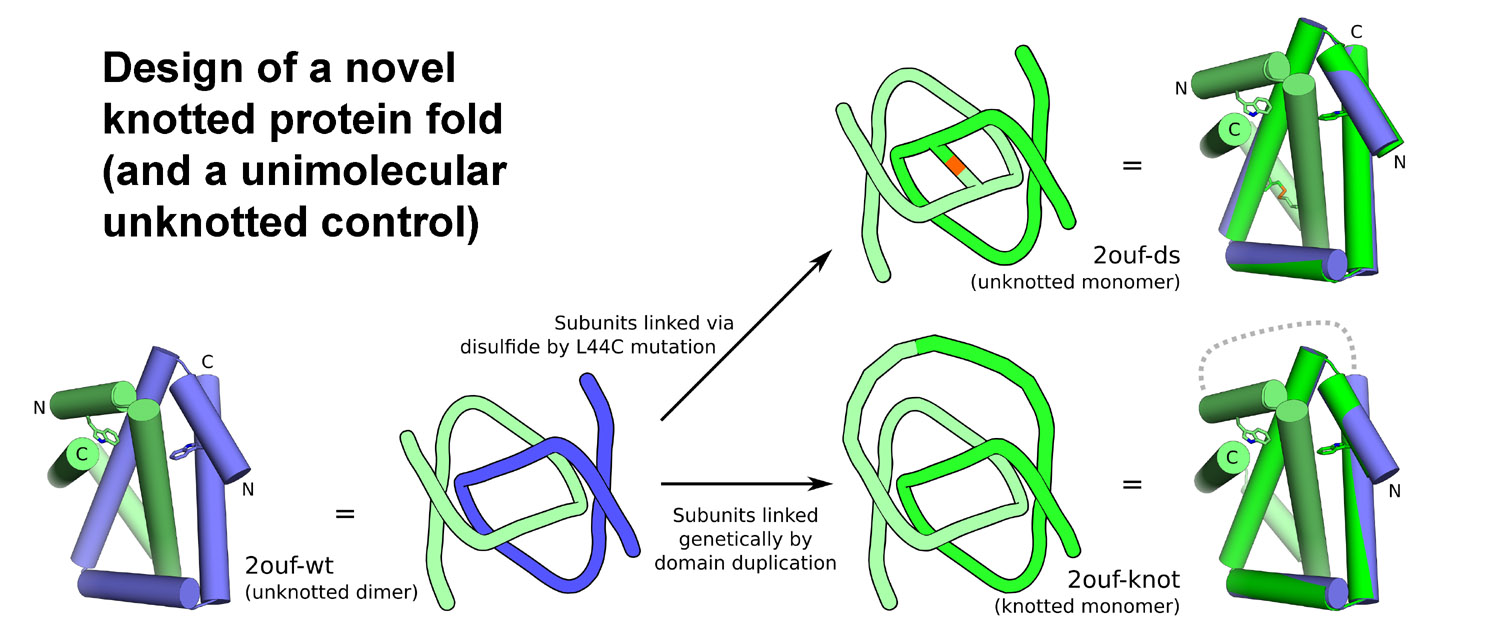
In subsequent work we used protein design to generate novel types of protein topologies
in order to test the consequences of topological complexity on protein
folding, and as a potential avenue for creating interesting types
of protein-based materials.
References:
- Sayre TC, Lee TM, King NP, Yeates TO. (2011).
Protein stabilization in a highly knotted protein polymer.
Protein Eng. Des. Sel.. Aug 2011. 24(8):627-30.
[Abstract]
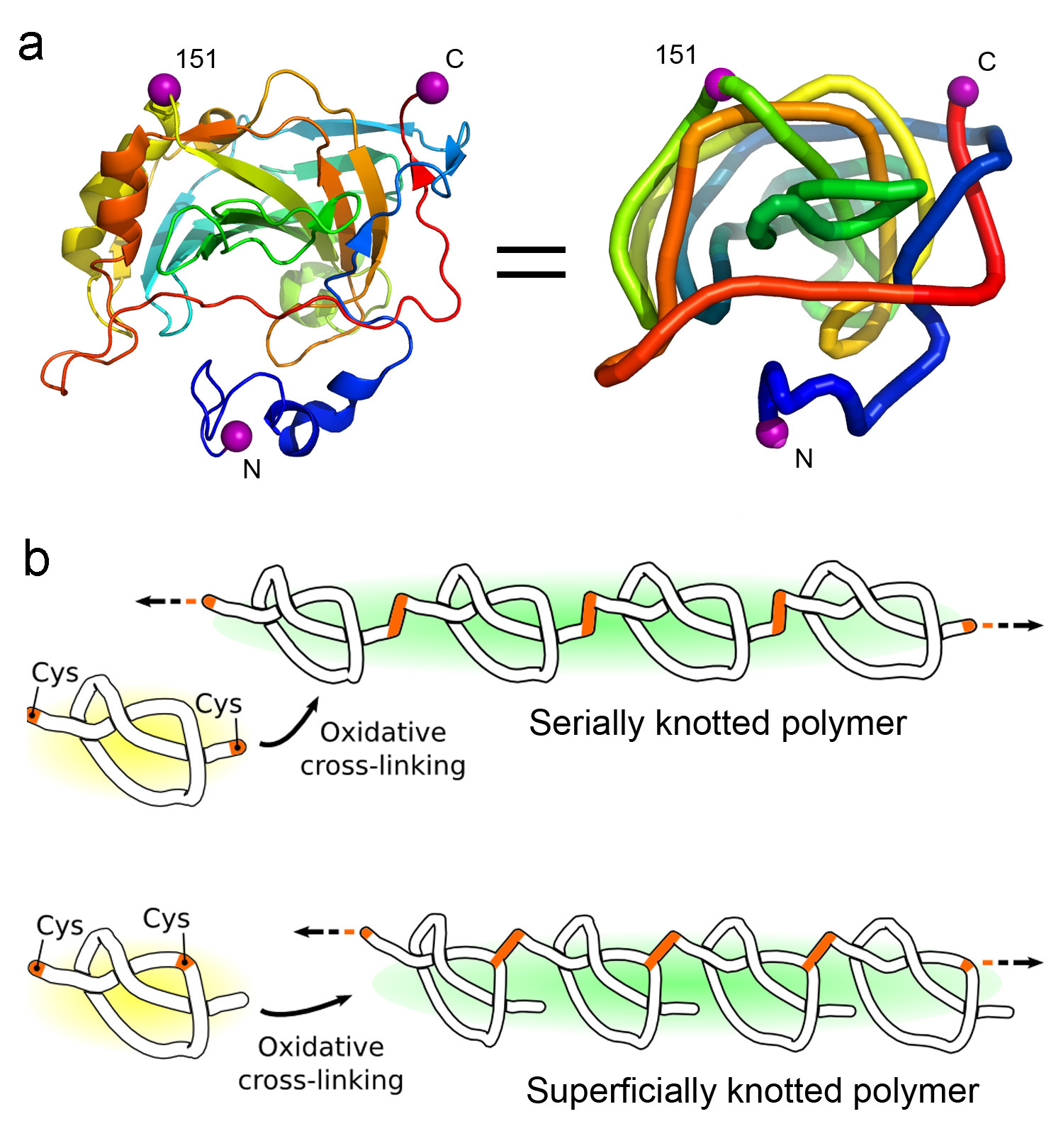
The polypeptide backbones of a few proteins are tied in a knot. The biophysical effects and potential biological roles of knots are not well understood. Here, we test the consequences of protein knotting by taking a monomeric protein, carbonic anhydrase II, whose native structure contains a shallow knot, and polymerizing it end-to-end to form a deeply and multiply knotted polymeric filament. Thermal stability experiments show that the polymer is stabilized against loss of structure and aggregation by the presence of deep knots.
- King NP, Jacobitz AW, Sawaya MR, Goldschmidt L, Yeates TO. (2010).
Structure and folding of a designed knotted protein.
Proc. Natl. Acad. Sci. U.S.A.. Nov 2010. 107(48):20732-7.
[Abstract]
A very small number of natural proteins have folded configurations in which the polypeptide backbone is knotted. Relatively little is known about the folding energy landscapes of such proteins, or how they have evolved. We explore those questions here by designing a unique knotted protein structure. Biophysical characterization and X-ray crystal structure determination show that the designed protein folds to the intended configuration, tying itself in a knot in the process, and that it folds reversibly. The protein folds to its native, knotted configuration approximately 20 times more slowly than a control protein, which was designed to have a similar tertiary structure but to be unknotted. Preliminary kinetic experiments suggest a complicated folding mechanism, providing opportunities for further characterization. The findings illustrate a situation where a protein is able to successfully traverse a complex folding energy landscape, though the amino acid sequence of the protein has not been subjected to evolutionary pressure for that ability. The success of the design strategy--connecting two monomers of an intertwined homodimer into a single protein chain--supports a model for evolution of knotted structures via gene duplication.
- King NP, Yeates EO, Yeates TO. (2007).
Identification of rare slipknots in proteins and their implications for stability and folding.
J. Mol. Biol.. Oct 2007. 373(1):153-66.
[Abstract]
Among the thousands of known three-dimensional protein folds, only a few have been found whose backbones are in knotted configurations. The rarity of knotted proteins has important implications for how natural proteins reach their natively folded states. Proteins with such unusual features offer unique opportunities for studying the relationships between structure, folding, and stability. Here we report the identification of a unique slipknot feature in the fold of a well-known thermostable protein, alkaline phosphatase. A slipknot is created when a knot is formed by part of a protein chain, after which the backbone doubles back so that the entire structure becomes unknotted in a mathematical sense. Slipknots are therefore not detected by computational tests that look for knots in complete protein structures. A computational survey looking specifically for slipknots in the Protein Data Bank reveals a few other instances in addition to alkaline phosphatase. Unexpected similarities are noted among some of the proteins identified. In addition, two transmembrane proteins are found to contain slipknots. Finally, mutagenesis experiments on alkaline phosphatase are used to probe the contribution the slipknot feature makes to thermal stability. The trends and conserved features observed in these proteins provide new insights into mechanisms of protein folding and stability.
- Yeates TO, Norcross TS, King NP. (2007).
Knotted and topologically complex proteins as models for studying folding and stability.
Curr Opin Chem Biol. Dec 2007. 11(6):595-603.
[Abstract]
Among proteins of known three-dimensional structure, only a few possess complex topological features such as knotted or interlinked (catenated) protein backbones. Such unusual proteins offer potentially unique insights into folding pathways and stabilization mechanisms. They also present special challenges for both theorists and computational scientists interested in understanding and predicting protein-folding behavior. Here, we review complex topological features in proteins with a focus on recent progress on the identification and characterization of knotted and interlinked protein systems. Also, an approach is described for designing an expanded set of knotted proteins.
- Norcross TS, Yeates TO. (2006).
A framework for describing topological frustration in models of protein folding.
J. Mol. Biol.. Sep 2006. 362(3):605-21.
[Abstract]
In a natively folded protein of moderate or larger size, the protein backbone may weave through itself in complex ways, raising questions about what sequence of events might have to occur in order for the protein to reach its native configuration from the unfolded state. A mathematical framework is presented here for describing the notion of a topological folding barrier, which occurs when a protein chain must pass through a hole or opening, formed by other regions of the protein structure. Different folding pathways encounter different numbers of such barriers and therefore different degrees of frustration. A dynamic programming algorithm finds the optimal theoretical folding path and minimal degree of frustration for a protein based on its natively folded configuration. Calculations over a database of protein structures provide insights into questions such as whether the path of minimal frustration might tend to favor folding from one or from many sites of folding nucleation, or whether proteins favor folding around the N terminus, thereby providing support for the hypothesis that proteins fold co-translationally. The computational methods are applied to a multi-disulfide bonded protein, with computational findings that are consistent with the experimentally observed folding pathway. Attention is drawn to certain complex protein folds for which the computational method suggests there may be a preferred site of nucleation or where folding is likely to proceed through a relatively well-defined pathway or intermediate. The computational analyses lead to testable models for protein folding.
|
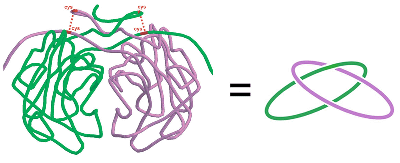
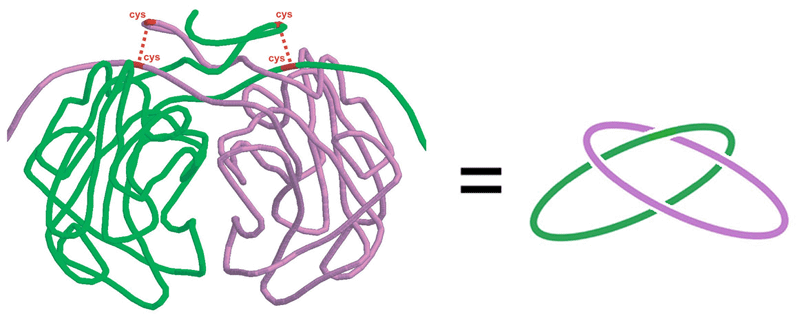
A covalently and topologically linked dimer, citrate synthase, in the hyperthermophile P. aerophilum. (Adapted from Boutz, et al.)
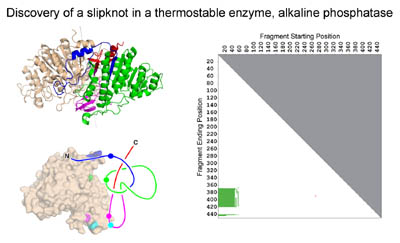
Discovery of the first protein slip knot, in the thermophilic enzyme alkaline phosphatase. The panel on the right shows a knot plot that revealed that a partial structure within the intact fold forms a knot. (Adapted from King, et al. (2007))
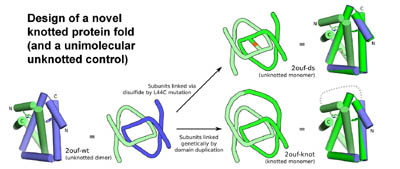
Design of a novel knotted protein fold. Protein folding experiments demonstrate that the knot substantially complicates the folding landscape and retards folding. (Adapted from King, et al. 2010).
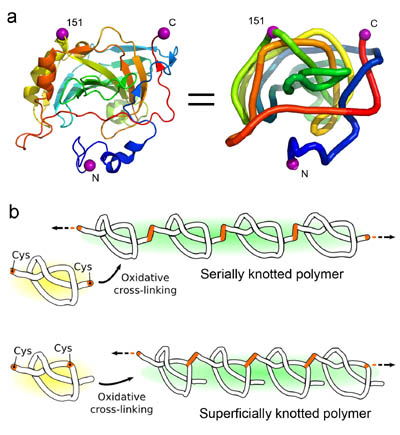
Polymerizing a shallowly knotted protein produces a series of deep knots, which stabilizes the protein compared to the control. (Adapted from Sayre, et al.)
|