|
By the early 1990's it had become clear that it was possible to
build and refine atomic structures that had reasonably good
crystallographic R-values, but which contained serious
structural errors.
This motivated the development of programs for evaluating the
correctness of structures that were either in the process of atomic refinement,
or which had already been deposited into the PDB.
Among the first was an approach by Luthy and Bowie in David Eisenberg's
group (Luthy, Bowie, and Eisenberg (1992) Nature 356, 83-85),
named Verify3D, which assessed the degree to which the environment
(polarity for example) around each amino acid in a structure was
statistically consistent with the amino acid type at that position.
Shortly thereafter, other programs were developed that took the analysis
from the level of amino acid residues to the atomic level. One of
those was ERRAT, written by Chris Colovos as an undergraduate in
the Yeates group (Colovos and Yeates (1993)
Verification of protein structures: patterns of non-bonded atomic interactions.
Protein Sci. 2, 1511-1519). The ERRAT program classifies atoms into
three types (C, N, and O) and asks whether the distribution of non-bonded
interactions between atoms in a candidate structure matches the distribution
established from a database of reliable, high resolution structures.
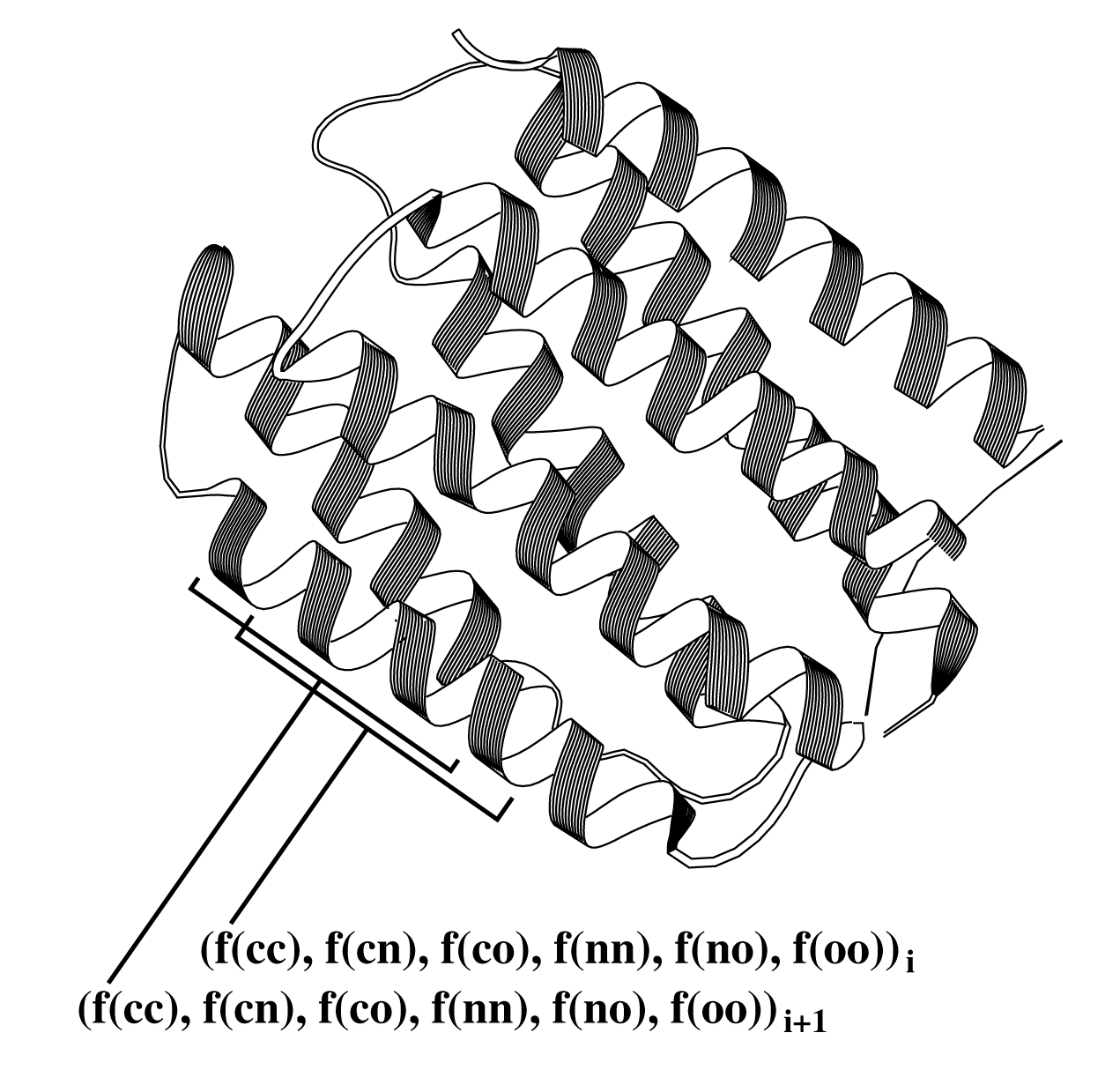
A structure is evaluated in a 9-residue sliding window.
For each such window, the number of interactions of the 6 possible
types (CC,CN,CO,NN,NO,OO) is totaled. These six counts are then converted
to fractional values by dividing by the total number of interactions.
Because those six normalized values sum to unity, the values span a five
dimensional space. The distribution of interaction frequencies for
correct structures can therefore be
characterized as a generalized Gaussian function in 5-dimensional space.
Based on this distribution, the interaction frequencies observed for
a candidate structure can be evaluated for the likelihood that they
could have been drawn at random from the correct distribution.
For each 9-residue window, the atomic interactions tabulated are all
those that involve at least one atom from that
window, and which are less than a distance cutoff of 3.5A.
The ERRAT program was found to be very effective in
identifying erroneous regions of model structures, and to be particularly
useful during the process of building and refining crystal structures. One
weakness of the program was a high sensitivity to small deviations in
atomic positions, owing mainly to the discontinuous nature of the error
function arising from the distance cutoff. Around 2002, another undergraduate
student Dennis Obukov, rewrote ERRAT with a continuous distance
weighting scheme, which led to more stable and robust behavior.
That version of ERRAT replaced the original one and remains accessible
through a web server at UCLA. Recent incarnations
of statistical or knowledge-based
atomic-level energy functions for evaluating protein structures
include Zhou's program DFIRE.
|
 |