|
Structural Molecular Biology Laboratory, CHEMM230D |
|
|

|
1) Getting the most from your amino acid sequence. 2) Concentrating your protein. 3) Hanging drop vapour diffusion method of crystallization explained. 4) A Practical Guide to Crystallization by Mark Knapp.
|
|
|
There are several important questions the crystallographer should ask about a protein before beginning a crystallization experiment. What is the moleular weight of the protein? What buffer will the protein be stable in? Will the protein remain active if I boil it in concentrated HCl? In the early days of crystallography, these questions could be directed to the biochemist who had painstakingly characterized the protein in full detail for many years in advance of structural studies. Nowadays, with the advent of structural genomics projects, the amino acid sequence may be the only information a crystallographer has to go on. In this section, we explore how many of these important questions can be answered on the internet given only the amino acid sequence.
The amino acid sequence of Proteinase K from Tritirachium album
GAAQTNAPWGLARISSTSPGTSTYYYDESAGQGSCVYVIDTGIEASHPEF
EGRAQMVKTYYYSSRDGNGHGTHCAGTVGSRTYGVAKKTQLFGVKVLDDN
GSGQYSTIIAGMDFVASDKNNRNCPKGVVASLSLGGGYSSSVNSAAARLQ
SSGVMVAVAAGNNNADARNYSPASEPSVCTVGASDRYDRRSSFSNYGSVL
DIFGPGTSILSTWIGGSTRSISGTSMATPHVAGLAAYLMTLGKTTAASAC
RYIADTANKGDLSNIPFGTVNLLAYNNYQA
Answer the questions below by pasting the sequence of proteinase K in the web site given below.
| Question | Web site |
| What is the molecular weight of my protein? Larger proteins often have multiple floppy domains which make the protein difficult to crystallize. Single domain proteins are usually less than 30 kDa. | http://ca.expasy.org/tools/protparam.html |
| How many amino acids are in the protein? How many are methionine? Generally, more than one methionine per 100 amino acids will ensure the success of a selenomet MAD experiment. | http://ca.expasy.org/tools/protparam.html |
| Are there any tryptophan residues in the protein sequence? Tryptophans are good points of reference for assigning sequence to electron density maps because of their large size and distinctive shape. But more importantly, the presenece of tryptophans endow a significant extinction coefficient at 280 nm wavelength that facilitate protein concentration determination by spectrophotometry. (Tyr, Phe, and disulfides also contribute a minor component to the extinction coefficient, but in the absence of tryptophan, their contribution may not be enough to allow accurate spectrophotometric determination of protein concentration). | http://ca.expasy.org/tools/protparam.html |
| What is the extinction coefficient at 280 nm? A valuable number for determining protein concentration by spectrophotometry. The Biuret or BioRad assays are alternative methods of determining protein concentration, but they are more time consuming. | http://ca.expasy.org/tools/protparam.html |
| Are there any free cystein residues? Cysteins are reactive with mercury and other class B heavy metal ions. Their presence suggest that mercury could be used as a derivative for phasing. One should also consider the cellular location of the protein. If it is extracellular, the cysteins could be oxidized in a disulfide bond, reducing cystein's reactivity toward heavy atoms. | http://ca.expasy.org/tools/protparam.html |
| What is the pI of my protein? The pI will determine which way the protein will migrate in an electric field in a native gel band-shift assay for potential heavy atom derivatives. Also, if the protein is insoluble when concentrated, one should consider using a buffer that is farther away from the pI. | http://ca.expasy.org/tools/protparam.html |
| Are there any homologs to my protein? What are their functions? A close homolog in the PDB (better than 30% sequence similarity) will allow phasing by molecular replacement. Also, knowledge of a homolog's biochemistry can be used to fill in gaps in knowledge about the subject protein if it is not well studied. | http://www.ncbi.nlm.nih.gov/BLAST/ (use blastp) |
| Are there any ligands that I can complex with my protein to help stabilize the protein conformational flexibility? The more rigid a protein, the more likely it will crystallize. Furthermore, a complex with substrate or substrate analog is inherently more scientifically interesting and revealing. If there is literature existing for your protein, you can search for it by keyword on PUBMED. If no literature exists for your protein, you can search for literature about the homolog that you discovered in the blast search above. | http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=PubMed |
| What is the secondary structure prediction for my protein? It is helpful to know secondary structure prediction when you are judging the quality of your first experimental electron density map. If the prediction suggests all helices and you can't find a single helix in the map, then you may call into question the quality of your map? | http://bioinf.cs.ucl.ac.uk/psipred/psiform.html many other options listed by expasy web site. |
|
|
Crystallographers are THE experts in protein concentration. Few other experiments in biochemistry require as high concentrations of pure protein as crystallization experiments. Crystallization typically requires 10 mg/mL concentration for proteins in the 10-30 kDa range. Larger protein tend to require less concentration (2-5 mg/mL). Smaller proteins tend to require higher concentration (20-50 mg/mL). There is a kit available to help you test whether your protein is at a suitable concentration for initial crystallization trials. It contains several precipitating agents commonly used in crystallization. If upon addition of these agents to 3 uL of your protein you find precipitation forming, then it is likely that your protein concentration is appropriate for crystallization trials. To get the Crystool Pre-Screen kit (free?), send e-mail to Bernhard Rupp.
 I typically concentrate protein
using a Centricon
concentrator
from Amicon. The centricon is designed to use centrifugal force to push
solvent out through pores in a membrane, while your protein retained inside
by size exclusion. The centricon holds up to 2.5 mL and concentrates down
to a minimum of about 100 uL in an SS34 rotor. Concentration requires about
2 hours at 5000 rpm.
I typically concentrate protein
using a Centricon
concentrator
from Amicon. The centricon is designed to use centrifugal force to push
solvent out through pores in a membrane, while your protein retained inside
by size exclusion. The centricon holds up to 2.5 mL and concentrates down
to a minimum of about 100 uL in an SS34 rotor. Concentration requires about
2 hours at 5000 rpm.
ALWAYS check the concentration of the protein before you use a centricon. Good recovery from a centricon is not always assured in the beginning, so you want to be able to check that you get the same amount of protein out of the centricon that you put in. Good recovery depends on chosing the correct molecular weight cutoff (MWCO), and the proper buffer: There are different MWCOs (3kDa, 10kDa, 30kDa) for different size proteins. The larger the MWCO, the faster the concentration process. Be careful to chose a MWCO that is significantly smaller than your protein's molecular weight. Otherwise, the protein may get trapped in the membrane pores. It is also a good idea to include at least 10 mM NaCl to prevent the protein from sticking to the surface of the membrane. If the ionic strength is too low, the hydrophobic parts of the protein glom onto the membrane surface, blocking the pores and slowing down the concentration process. When this phenomenon occurs, you could also significantly reduce the yield of protein recovery.
Achieving an adequate protein concentration while maintaining solubility
can be a tricky problem for some proteins. You may find that your protein
"oils out" of solution. (i.e. You find a clear jelly at the bottom of the
centricon recovery tube.) Or perhaps it precipitates in a white powdery
cake. These things happen because the protein would rather interact with
another protein molecule than with solvent molecules. The solution to the
"non-solution" usually lies in finding the correct buffer pH or chemical
additive. Change the pH of the buffer further away from the pI of the protein
in order to increase the charge on the protein and the favorability of
its interaction with solvent. Also, the addition of small polar organic
molecules such as glycerol,
sucrose, methylpentanediol, or 1,6-hexanediol often dissolves protein oils
instantly. If you know that your protein uses metal ions, or binds a ligand,
the addition of these chemicals could also improve protein solubility.
In very difficult cases, you could use a mild detergent such as
beta-octyl
glucoside. A convenient way to test different compounds for their solubilizing
effect on the protein is to take a drop of protein oil or precipitate on
a glass coverslip and add some chemical to it while watching the drop in
the microscope. If the oil or precipitate dissolves, then you can add that
chemical to your protein stock before concentration to maintain solubility.
|
|
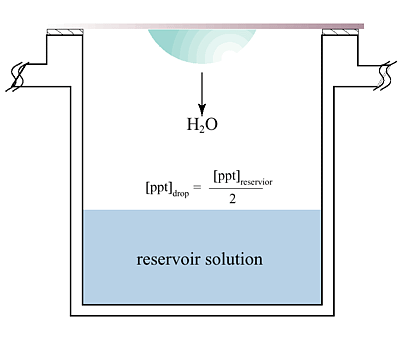 The hanging
drop vapor diffusion technique is the most popular method for the crys
tallization of macromolecules. The principle of vapor diffusion is straightforward.
A drop composed of a mixture of sample and reagent is placed in vapor equilibration
with a liquid reservoir of reagent. Typically the drop contains a lower
reagent concentration than the reservoir. To achieve equilibrium, water
vapor leaves the drop and eventually ends up in the reservoir. As water
leaves the drop, the sample undergoes an increase in relative supersaturation.
Both the sample and reagent increase in concentration as water leaves the
drop for the reservoir. Equilibration is reached when the reagent concentration
in the drop is approximately the same as that in the reservoir.
The hanging
drop vapor diffusion technique is the most popular method for the crys
tallization of macromolecules. The principle of vapor diffusion is straightforward.
A drop composed of a mixture of sample and reagent is placed in vapor equilibration
with a liquid reservoir of reagent. Typically the drop contains a lower
reagent concentration than the reservoir. To achieve equilibrium, water
vapor leaves the drop and eventually ends up in the reservoir. As water
leaves the drop, the sample undergoes an increase in relative supersaturation.
Both the sample and reagent increase in concentration as water leaves the
drop for the reservoir. Equilibration is reached when the reagent concentration
in the drop is approximately the same as that in the reservoir.
See also other selected readings from Hampton Research.
For a thorough, well written article about current approaches to macromolecular
crystallization, see Current Approaches to macromolecular Crystallization
by Alexander McPherson, Eur. J. Biochem. 189, 1-23 (1990).
You can get a copy from Mike.