Refinement/Structure Validation
Structural Molecular Biology Laboratory, ChemM230D
|
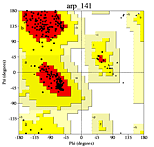
Ramachandran plot from a high quality model.
|
Suggested Reading Materials
1) Electron
density map intepretation by T. A. Jones and M. Kjeldgaard, Methods in
Enzymology, Vol 277 (1997) pages 173-207. This link is a 17 page PDF
file
2) Refmac5
manual by Garib Murshudov, University of York and CLRC, Daresbury Laboratory
and Alexei Vagin, Martyn Winn, Roberto Steiner, Eleanor Dodson, Maria Turkenburg,
Kim Henrick, Liz Potterton.
3)Verification of Protein Structures:
Patterns of Nonbonded Atomic Interactions by C. Colovos & T. O. Yeates.
Protein Science (1993), 2, 1511-1519. Describes ERRAT program.
4) Assessment of protein models with three-dimensional
profiles by Roland. Luthy, James U. Bowie, & David Eisenberg. Nature
(1992), 356, 83-85.
5) powerpoint presentation
6) Quality Control Server
7) UCLA Saves Server
|
5th Assignment:
Structure
Refinement Table
due at the
end of lab period. |
Illustrations
|
| Objective: To produce a crystallographic
structure refinement table typically found in structural biology journals.
Method: Copy the format of the table on
the left. Substitute the data from your pdb file for the data in the
table.
|
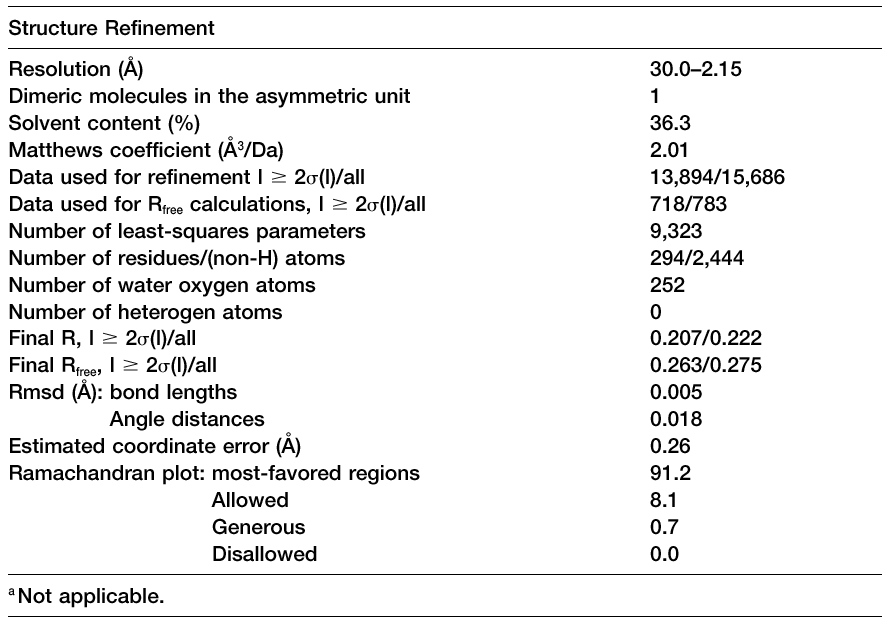
A typical Table 3, adapted from Blaszczyk et al.,
Crystallographic and Modeling Studies of RNase III Suggest a Mechanism
for
Double-Stranded RNA Cleavage, Structure, Vol. 9,
1225-1236, December 2001. |
|
|
| Part One: Refinement, difference Fourier map calculation, and model
building. |
Illustrations
|
| Objective: To
refine the PMSF complex structure and model the PMSF inhibitor in the Fo-Fc
electron density map.
Strategy: Use the coordinates of the native
proteinase K structure output by ARP/wARP as a starting point for the refinement
of the PMSF complex. Refine using maximum likelihood restrained refinement
for 5 cycles. Calculate difference Fourier maps (fo-fc and 2fo-fc) and look
for large peaks in the fo-fc density. One of these peaks should correspond
to PMSF, the others may be due to water molecules, side chain movements, or
bits of the model that were not built by ARP/wARP previously.
Procedures: Login to Bernal, Laue, or Bragg.
Change directories to your working directory. Type the word "ccp4i" to start
the CCP4 programs GUI. Define yourself as the user in the "Directories &
Projects Dir" button. Select the options as shown in the example on
the right. Check the R-work and R-free. Begin an O session
by typing "ono" and in the graphic window type "@difmacro".
This command will read in the refined proteinase K model and a few libraries
that will aid in model building. Tear off the following sub-menus form
the "menus" menu: Objects, User, Fake Dials. You should be able to see
three objects: the proteinase K model (prok1); the 2fo-fc difference Fourier
map (2fofc); and the fo-fc difference Fourier map contoured in positive(pos/blue)
and negative (neg/red) contour levels. You will see a sidechain movement
near Trp A9; fix it with RSR-rotamer. You will see a missing dipeptide between
A171-A174 (why would this be missing from the model?); build it in with "build_resi
prok1 a172 f" followed by "build_resi prok1 a173 f". Adjust the conformation
by "grab residue". Lastly, you will see some density for the PMSF.
Read in pms.pdb and position it near the active site serine. Write
out coordinates for your modified protein (prok_pmsf1.pdb) and coordinates
for you pmsf molecule (pms.pdb). Catenate the pmsf coordinates to the
bottom of prok_pmsf1.pdb and use this file for the next round of refinement.
|
 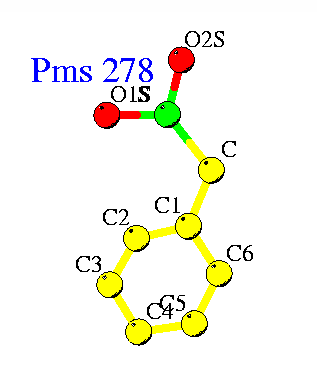
PMSF binds to active site serine residue
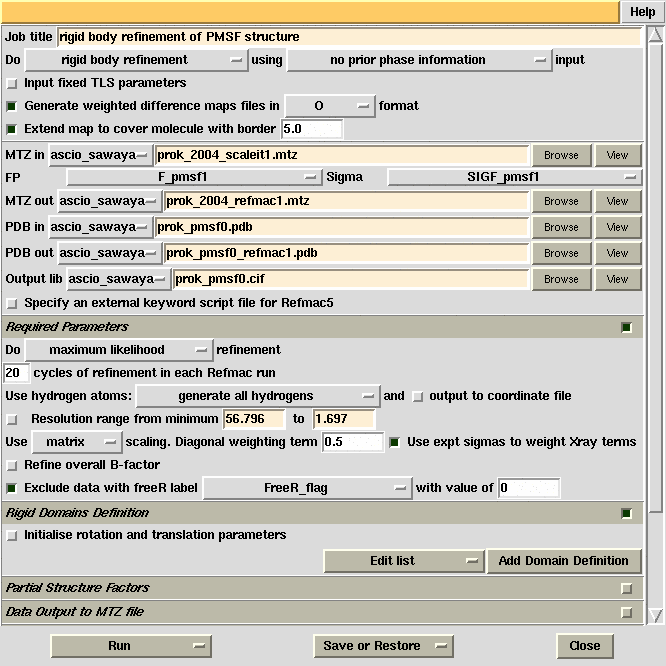
Refmac5 GUI used for initial refinement of the proteinase
K structure against the PMSF complex data and produce difference Fourier maps.
|
| Part Two: Refine your PMSF complex |
Illustrations
|
| Objective: To refine the PMSF that
you have modeled.
|
|
Procedures: Refine using the PMS dictionary
file and the model containing pmsf coordinates.
Mike's spiel.
|
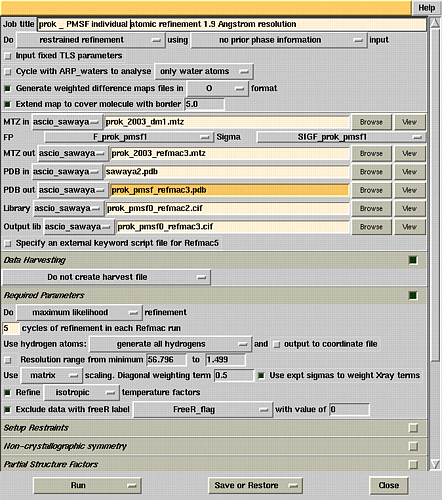
Refmac5 refinement window.
|
| |
|
| Part Three: Structure vaildation. |
Illustrations
|
| Objective: Check the quality
of your model with Ramachandran,Errat, and Verify3D
plots.
Procedures:
Run procheck,
errat, verify3D or just
do it all with the SAVS
server.
|
001_MAAQTNAPWG_LARISSTSPG_TSTYYYDESA_GQGSCVYVID
041_TGIEASHPEF_EGRAQMVKTY_YYSSRDGNGH_GTHCAGTVGS
081_RTYGVAKKTQ_LFGVKVLDDN_GSGQYSTIIA_GMDFVASDKN
121_NRNCPKGVVA_SLSLGGGYSS_SVNSAAARLQ_SSGVMVAVAA
161_GNNNADARNY_SPASEPSVCT_VGASDRYDRR_SSFSNYGSVL
201_DIFGPGTSIL_STWIGGSTRS_ISGTSMATPH_VAGLAAYLMT
241_LGKTTAASAC_RYIADTANKG_DLSNIPFGTV_NLLAYNNYQA
|
|
|
|
|
|
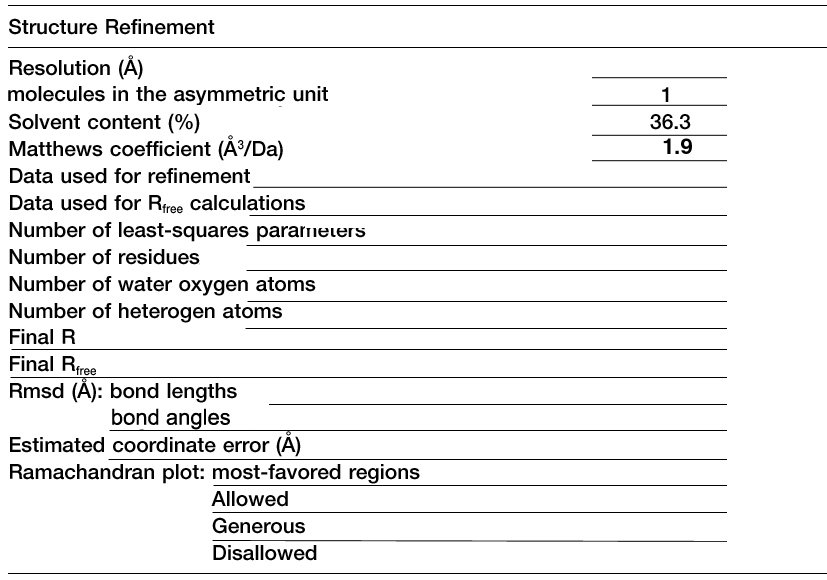
Fill in the blanks with Proteinase K data listed in your PDB file.
Instructor's
preparations
Back to CHEM
M230D course syllabus
[Overview] ·[Facilities] ·
[People] · [Services]
·[Lectures] ·
[BioLinks] ·
[Stats] ·[Search]
|
|
|