|
|
|
|
 |
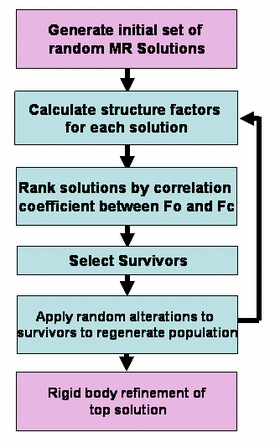
EPMR is an efficient six-dimensional search carried out using
an evolutionary optimization algorithm. In this procedure, a population of
initially random molecular replacement solutions is iteratively optimized
with respect to the correlation coefficient between observed and calculated
structure factors. The sensitivity and reliability of the method is enhanced
by uniform sampling of the rotational search space and the use of continuously
variable rotational and translational parameters. The process is several
orders of magnitude faster than a systematic six-dimensional search, and
comparisons show that it can identify solutions using significantly less
accurate or less complete search models than is possible with two existing
molecular-replacement methods.
Manual for EPMR: openEPMR v0.2 manual (open source) Reference for EPMR: |
STEP ONE
|
|
|
||||||||||||||
| #!/bin/csh
-f source /joule2/programs/login cd /directory/where/this/command/file/exists epmr -m1 -h4 -l15 -n50 -t1 -b25 cell.dat model.pdb data.fin >epmr.log EPMR Algorithm in a nutshell
 |
The first line is to declare c shell. Second line sets up the environment variable that tells the computer where the epmr program is located. Third line specifies the location of this command file. It is necessary only when submitting to the batch queue. Fourth line, the epmr command line (bottom), should be a single continuous line. It may look like two lines here because of wraparound.
|
STEP TWO
|
|
|
|
for plotting correlation coefficients
grep CC epmr.log | awk '{printf("%8.3f \n" ,$12 );}' > cc.logxmgr cc.log for plotting R-factors
grep CC epmr.log | awk '{printf("%8.3f \n" ,$14 );}' > r.logxmgr r.log EXAMPLES
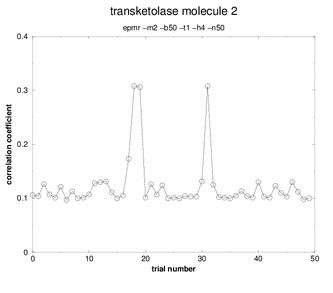 Peaks! Three of the solutions stand out above
all the others. Any of these three solutions would be correct.
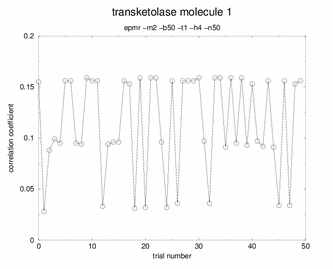 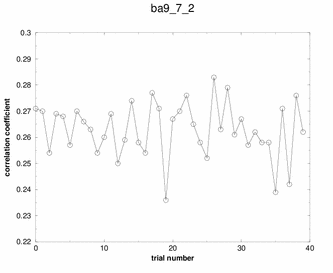 Booh! Hisssss! No spikes, no plateaus. Just random garbage. |
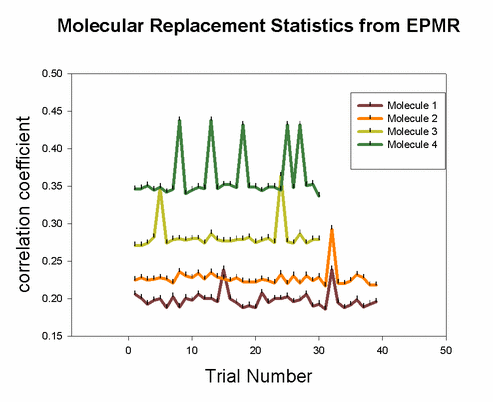
Although many potential solutions are calculated, EPMR will output only the solution with the highest correlation coefficient (unless otherwise specified). It is wise to check what its correlation coefficient is. You would hope that for a correlation coefficient over 35%. But, the ultimate test of whether the solution is correct is to demonstrate that you can refine the structure with a concommitant drop in the free R factor. During the EPMR calculation, one may wish to check the progress by plotting the correlation coefficient (CC) as a function of the trial number. This simple script on the left will extract from epmr.log a list of the correlation coefficients and put them into a file called cc.log. You may plot the file by typing "xmgr cc.log". You will get a graph like the graphs shown on the left. It is always encouraging to see a spike in the graph. But this is not a necessary feature for a correct solution. What do I do if EPMR does not produce a solution? You could try running the program with different resolution ranges. Instead of 15-4Ang, try 15-5Ang. You could also try more runs (-n100). Or try the conventional rotation/translation functions. A different algorithm might do the trick.
|