|
|
|
|
|
|
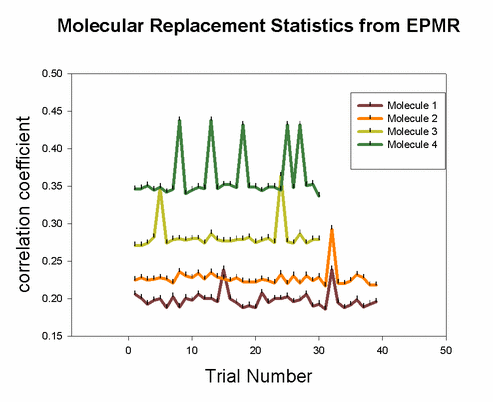
Pros: 1) Quick and easy to set up. Just a single line command script. Often successful. Of all the molecular replacement programs available, I usually start with EPMR first because it is so easy to do, and it takes a long time to run. So you can investigate other molecular replacement programs as it is running. 2) Works well with multiple molecules in asymmetric unit, if the model is very accurate. A critical factor in achieving success is the inclusion of a bump distance constraint that penalizes potential solutions if atoms overlap. Why? because two objects cannot occupy the same place at the same time. 3) Claimed by authors in manual: As long as the true solution represents the global maximum in the correlation coefficient between Fo and Fc, even if by the slimmest of margins, EPMR will eventually find it. 4) Works on a DEC Alpha station or linux. Fastest speed when submitted to linux cluster queue. 5) Runs a few cycles of rigid body refinement for each trial, so you dont have to do it later. Cons: References: Charles R. Kissinger, Daniel K. Gehlhaar & David B. Fogel (1999) "Rapid automated molecular replacement by evolutionary search", Acta Crystallographica, D55, 484-491. |
|
|
1) It is easy to use. Requires only a few scripts. 2) it can be run directly on Scalepack output (or any ascii format) 3) coordinate system conventions can be explicitly chosen 4) the option of origin removal is provided 5) the option of fast or slow rotation functions is provided 6) the output is easy to interpret; relevant parameters are conveniently printed on each plot. 7) The translation function results give TF peak height, CC, R-factor, and packing analysis unlike many other programs. 8) It is part of the "Replace" suite of programs and so provides a direct route to the locked rotation and translation functions which aids molecular replacement. Cons: References: L. Tong & M. G. Rossmann, Methods in Enzymology 276, 594-611, 1997.
|
| AMoRe
|
AMoRe Pros: The value of a heavy atom derivative can be more accurately judged from the appearance of a difference Patterson map -- this could be an isomorphous difference Patterson map (i.e. using differences in intensities between a native crystal and a heavy atom derivative) or an anomalous difference Patterson map (i.e. using differences in intensities between Bijvoet pairs). Planning to solve your MIR structure automatically with SOLVE? Check for peaks on your isomorphous difference Patterson first. If you have significant peaks on your Harker section (greater than 4 sigma), then SOLVE is likely to work. If not, then move on to the next derivative. If you have identified heavy atom sites by means of SOLVE or SHELXD, You should check the difference Patterson to see whether all the sites identified are correct. Through the use of xpatpred we see site 1 has been identified correctly on the figure at left. Requirements: Unit cell parameters, space group, and observed structure factors in fin format. References:M. G. Rossmann and D. M. Blow, Acta Cryst. 15, 24-31, 1962. |
| Locating
Heavy Atom Sites
|
There are several programs
which will locate heavy atom positions from difference Patterson maps: HASSP
(from the Heavy suite of programs and also utilized in SOLVE), Hercules (from
the XtalView suite of programs), Patsol (from Liang Tong), and rsps (from
the CCP4 suite of programs). However, in my opinion, use SHELXD first.
SHELXD is easy to use and the output is the most informative and easily interpretable of all programs. It has been used successfully in locating Br in MAD experiments where other programs failed. It claims to produce "more complete and precise solutions" by integrating both Patterson and direct methods. Requirements: Unit cell parameters, space group, and observed intensities in Scalepack format. References:M. G. Rossmann and D. M. Blow, Acta Cryst. 15, 24-31, 1962. |
|
|
There are many steps to
the phasing process. After the heavy atoms have been located 1) their positions
must be refined 2) the phases are calculated using both configurations of
heavy atoms related by a center of inversion 3) solvent flattening is used
to improve the phases for both maps 4) visual inspection of the map to chose
the correct hand 5) model building into the correct map. There are programs
that can do all these steps automatically (e.g. SOLVE and ELVES). Here we
describe the invidual steps used by these programs.
Requirements: Unit cell parameters, space group, and observed structure factors in Scalepack format for native and derivative, coordinates of heavy atom sites. References: |