|
|
|
|
| Preparing
a useful derivative
|
There are many
things to consider:
1) Safety: When preparing heavy atom derivatives, special
care must be taken to prevent poisoning/irradiating yourself or others.
Requirements: Caution when handling heavy atoms, a native gel (Phast system OK), a few microliters of concentrated protein, and various heavy atom solutions, crystals not necessary at this point. (hurray!) References:T. J. Boggon and L. Shapiro Structure 8, R143-R149, 2000. |
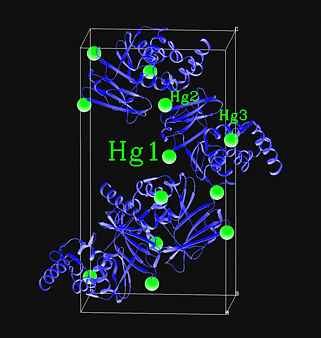
Evaluating Derivative Quality
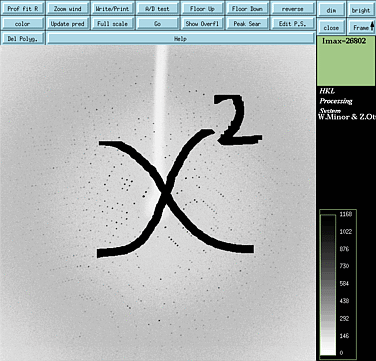 |
The quality of a heavy atom derivative
can be first evaluated by the chi squared test performed on either isomorphous
differences or anomalous differences. The chi squared test measures the
significance of the signal originating from the heavy atom. If the chi
squared statistics suggest that the differences are small, then the putative
derivative data is not useful for phasing. If the chi squared statistics
are large, then there is hope.
A positive chi squared test based on isomorphous differences is less reliable than a positive test based on anomalous differences. The poorer reliability of the isomorphous chi squared test is due to non specific heavy atom binding which often causes non-isomorphism between native and derivative crystals ...which in turn causes large chi squared values. Anomalous differences are measured from a single crystal (i.e. perfect isomorphism) so a positive chi squared test is more indicative of a true anomalous signal. Note that the anomalous signal is enhanced by chosing the peak wavelength. To find this wavelength, see the Anomalous Scattering Web Site Requirements: For isomorphous differences - a complete native data set in scalepack format (.sca file) and 5 degrees of integrated data (.x files) from a derivative data set. For anomalous differences - a highly redundant derivative data set in scalepack format (.sca) and processed with "anomalous" or "scale anomalous" flags in scalepack. |
| Difference Patterson Maps
Isomorphous OR Anomalous
|
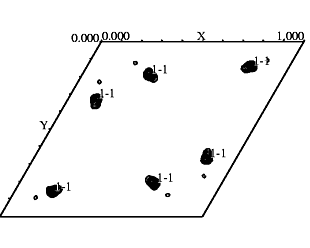
The value of a heavy atom derivative
can be more accurately judged from the appearance of a difference Patterson
map -- this could be an isomorphous difference Patterson map (i.e. using
differences in intensities between a native crystal and a heavy atom derivative)
or an anomalous difference Patterson map (i.e. using differences in intensities
between Bijvoet pairs). Planning to solve your MIR structure automatically
with SOLVE? Check for peaks on your isomorphous difference Patterson first.
If you have significant peaks on your Harker section (greater than 4 sigma),
then SOLVE is likely to work. If not, then move on to the next derivative.
If you have identified heavy atom sites by means of SOLVE
or SHELXD, You should check the difference Patterson to see whether all
the sites identified are correct. Through the use of xpatpred we see site
1 has been identified correctly on the figure at left.
List of
Harker Sections for all space groups.
Requirements: Unit cell parameters, space group,
and observed structure factors in fin format.
References:
Author, Journal volume, pages, year. |
| Locating
Heavy Atom Sites
|
There are several programs which will
locate heavy atom positions from difference Patterson maps: HASSP (from
the Heavy suite of programs and also utilized in SOLVE), Hercules (from
the XtalView suite of programs), Patsol (from Liang Tong), and rsps (from
the CCP4 suite of programs). However, in my opinion, use SHELXD first.
SHELXD is easy to use and the output is the most informative and easily interpretable of all programs. It has been used successfully in locating Br in MAD experiments where other programs failed. It claims to produce "more complete and precise solutions" by integrating both Patterson and direct methods. Requirements: Unit cell parameters, space group, and observed intensities in Scalepack format. References:M. G. Rossmann and D. M. Blow, Acta Cryst. 15, 24-31, 1962. |
|
or SIRAS phasing with Iodide
|
There are many steps to the phasing
process. After the heavy atoms have been located 1) their positions must
be refined 2) the phases are calculated using both configurations of heavy
atoms related by a center of inversion 3) solvent flattening is used to
improve the phases for both maps 4) visual inspection of the map to chose
the correct hand 5) model building into the correct map. There are programs
that can do all these steps automatically (e.g. SOLVE and ELVES). Here
we describe the invidual steps used by these programs. (1) for the
general MIRAS
case (multiple isomorphous replacment with anomalous scattering) and (2)
for SIRAS
phasing with iodide.
Requirements: Unit cell parameters, space group, and observed structure factors in Scalepack format for native and derivative, coordinates of heavy atom sites. References: |